
R語言 生存分析
阿新 • • 發佈:2018-11-08
文章目錄
R語言進行生存分析
1.下載示例資料
2.R語言程式碼例項詳解
#示例資料輸入 clinical <- read.table("clinical_clust.txt",header = T, row.names = 1, sep = "\t", check.names = F, na.strings = "", fill = T, stringsAsFactors = F) RPPA_Hierclust <- read.table("RPPA_Hierclust_clusters.txt", header = T, stringsAsFactors = F) #將兩個檔案需要的資料組合抽取並組合在一起 n_censor <- clinical[6,] clinical[6,which(n_censor == "DECEASED")] <- 0 clinical[6,which(n_censor != "DECEASED")] <- 1 clinical_json <- clinical[c(6, 3, 2, 1),] rownames(clinical_json) <- c("n_censor", "n_event", "surv", "time") colnames(clinical_json) <- NULL t_clinical_json <- t(clinical_json) t_clinical_json <- apply(t_clinical_json, 2, as.numeric) #將雙引號去除 t_surv <- t_clinical_json t_surv <- cbind(t_surv, as.matrix(RPPA_Hierclust$cluster)) colnames(t_surv)[5] <- "clust" #用R語言對上述資料進行生存分析 library(survival) #第一步:用Surv生成一個 survival object#### Sur_obj <- Surv(t_surv[,4], t_surv[,2]) #第一個引數是time,生存時間,對於右截尾資料,這是follow up time #第二個引數是event, 即the status indicator, normally 0=alive,1=dead #第二步:用survfit創造生存曲線模型#### model <- survfit(Sur_obj~1) #如果用所有的資料,不進行分組,則後面引數用1 model_1 <- survfit(Sur_obj~t_surv[,5]) #如果用聚類結果進行分組,則後面寫分組的結果 #第三步:用survdiff計算兩條或者多條生存曲線的差異顯著性#### km <- survdiff(Surv_obj~t_surv[,5]) #第四步:結果的形象化展示(結果展示見程式碼後面) plot(model) plot(model_2) #可以展示,但是結果美觀程度不夠 library(survminer) #用survminer進行漂亮的展示 ggsurvplot(model_2, main = "Survival curve", data = t_surv, pval=TRUE #新增P值 )
plot(model)圖片
plot(model_2)圖片
ggsurvplot(model_2) 圖片
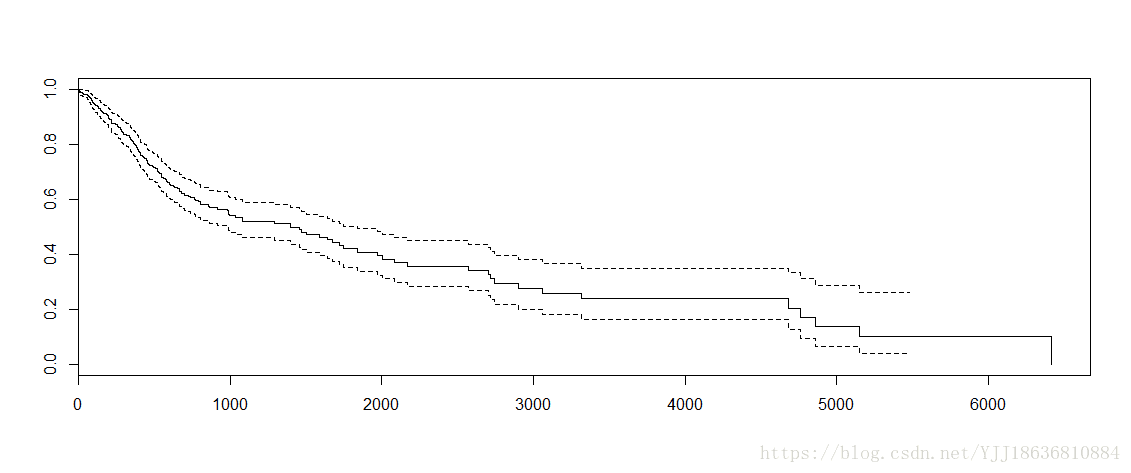
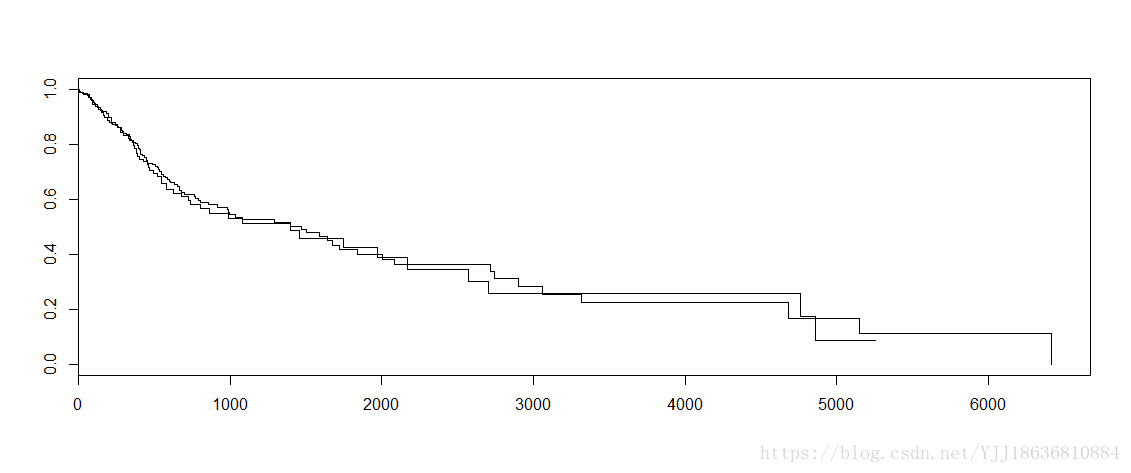
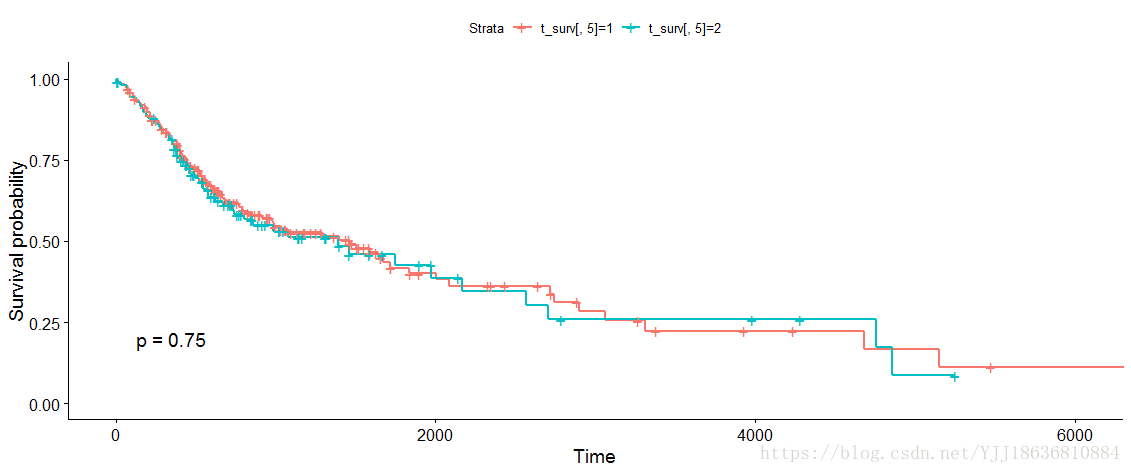
3.難點解讀
-
理解用R語言進行生存分析的大綱
(1)用Surv 生成survival object
(2)用survfit 生成擬合的生存曲線
(3)用survdiff 計算兩條或者兩條以上生存曲線的差異表達值
(4)用適當的方法展示生存曲線 -
P值雖然可以較好的展示出來,但是將P_value進行輸出並不容易,下面展示P_value 的輸出
km <- survdiff(Surv_obj~t_surv[,5]) p.value <- 1 - pchisq(km$chisq, length(km$n) - 1) #用chisq值和自由度的結果自行計算P.value print(p.value)
3.關於生存分析系列概念的理解,可以參考部落格:生存分析,裡面有非常詳細的講解
4.補充:如何用R語言 手動計算生存率
#延續上述資料輸入,將資料按照time進行排序,計算生存率並替換第三列的資料 #將surv行變成生存率 clust_unique <- unique(RPPA_Hierclust$cluster) clinical_clust <- NULL for (i in 1:length(clust_unique)){ i_station <- which(RPPA_Hierclust$cluster == i) t_clinical_json_clust <- t_clinical_json[i_station, ] #將t_clinical_json 按照time進行排序 unique_sort <- unique(sort(t_clinical_json_clust[,4])) right_sort <- NULL for(n in 1:length(unique_sort)){ right_sort <- c(right_sort, which(t_clinical_json_clust[,4] == unique_sort[n])) } t_clinical_json_clust <- t_clinical_json_clust[right_sort,] #將排序的後的資料計算生存率 allpatiants <- nrow(t_clinical_json_clust) unique_time <- unique(t_clinical_json_clust[,4]) P <- 1 n <- allpatiants for(j in 1:length(unique_time)){ time_position <- which(t_clinical_json_clust[,4] == unique_time[j]) censor_sum <- sum(t_clinical_json_clust[time_position, 1]) d <- sum(t_clinical_json_clust[time_position, 3]) P <- P*((n-d)/n) n <- n-d-censor_sum t_clinical_json_clust[time_position, 3] <- rep(P, length(time_position)) } #put it into clinical_clust clinical_clust[[i]] <- list(t_clinical_json_clust) }