
QIIME 2使用者文件. 4人體各部位微生物組分析實戰Moving Pictures(2018.11)
文章目錄
前情提要
QIIME 2使用者文件. 4人體各部位微生物組
“Moving Pictures” tutorial
https://docs.qiime2.org/2018.11/tutorials/moving-pictures/
注意:本文學習需要安裝好QIIME 2,請務必完成《1簡介和安裝》
在本教程中,你將使用QIIME 2在五個時間點對來自四個身體部位的兩個人的微生物組樣本進行分析,第一個時間點緊接著是抗生素的使用。基於這些樣本的研究文章《Moving pictures of the human microbiome》在2011年發表於Genome Biology
對於熟悉QIIME 1的使用者,本資料也出現在QIIME的教程中。
在開始本教程前,我們需要進入工作環境建立新目錄並進入
啟動QIIME2執行環境
對於上文提到了兩種常用安裝方法,我們每次在分析資料前,需要開啟工作環境,根據情況選擇對應的開啟方式。
# 建立qiime2學習目錄並進入 mkdir -p qiime2 cd qiime2 # Miniconda安裝的請執行如下命令載入工作環境 source activate qiime2-2018.11 # 如果是docker安裝的請執行如下命令,預設載入當前目錄至/data目錄 docker run --rm -v $(pwd):/data --name=qiime -it qiime2/core:2018.11 # 建立本節學習目錄 mkdir qiime2-moving-pictures-tutorial cd qiime2-moving-pictures-tutorial
樣本元資料
Sample metadata
在開始分析之前,探索樣本元資料,以熟悉本研究中使用的樣本。示例元資料作為Google 表格提供。你可以通過選擇 File > Download as > Tab-separated values,以製表符分隔的文字格式下載該檔案。或者,以下命令將作為製表符分隔的文字下載示例元資料,並將其儲存在檔案sample-metadata.tsv,這個sample-metadata.tsv檔案在本教程中一直被用到。
wget \
-O "sample-metadata.tsv" \
"https://data.qiime2.org/2018.11/tutorials/moving-pictures/sample_metadata.tsv"
注意:QIIME 2 官方測試資料均儲存在Google伺服器上,國內下載比較困難。可使用代理伺服器(如藍燈)下載此連結 https://data.qiime2.org/2018.11/tutorials/moving-pictures/sample_metadata.tsv ,或公眾號後臺回覆"qiime2"獲取測試資料批量下載連結,你還可以跳過以後的wget步驟。
提示:Keemei是一個用於驗證示例元資料的Google Sheets外掛。在開始任何分析之前,樣本元資料的驗證非常重要。嘗試按照Keemei網站上的說明安裝Keemei,然後驗證上面連結的示例元資料電子表格。該電子表格還包括一個帶有一些無效資料的表格,以便使用Keemei進行測試。
提示:要了解關於元資料的更多資訊,包括如何格式化元資料以便與QIIME 2一起使用,請參閱元資料教程。
下載和匯入資料
Obtaining and importing data
下載在本次分析中使用的序列。在本教程中,我們將處理完整的序列資料的一小部分,以便命令能夠快速執行(減少等待時間)。
建立子目錄並下載實驗測序資料,無法下載,請公眾號後臺回覆"qiime2"獲取測試資料備用下載連結。
mkdir -p emp-single-end-sequences
wget -O "emp-single-end-sequences/barcodes.fastq.gz" "https://data.qiime2.org/2018.11/tutorials/moving-pictures/emp-single-end-sequences/barcodes.fastq.gz"
wget -O "emp-single-end-sequences/sequences.fastq.gz" "https://data.qiime2.org/2018.11/tutorials/moving-pictures/emp-single-end-sequences/sequences.fastq.gz"
用於輸入到QIIME 2的所有資料都以QIIME 2物件的形式出現,其中包含有關資料型別和資料來源的資訊。因此,我們需要做的第一件事是將這些序列資料檔案匯入到QIIME 2物件中。
這個QIIME 2物件的語義型別是EMPSingleEndSequences。 QIIME 物件EMPSingleEndSequences是包含多樣本混合的序列檔案,這意味著序列尚未分配給樣本(因此包括sequences.fastq.gz和barcode.fastq.gz檔案,其中barcode.fastq.gz包含與sequences.fastq.gz中的每個序列相關聯的條形碼)。要匯入其他格式的序列資料,請參閱匯入資料教程。
匯入資料:生成qiime2要求的物件格式
qiime tools import \
--type EMPSingleEndSequences \
--input-path emp-single-end-sequences \
--output-path emp-single-end-sequences.qza
輸出物件:
emp-single-end-sequences.qza: 檢視 | 下載
注意:公眾號無法開啟外部連結,想要直接訪問檢視、下載連結,可訪問QIIME 2論壇、CSDN或科學網閱讀同名文件,可用百度搜索本節標題直達。
提示:
上面的檢視和下載由文件中的命令建立的QIIME 2物件和視覺化連結。例如,上面的命令建立了單個emp-single-end-sequences.qza檔案,上面連結了相應的預計算檔案(輸出結果)。你可以檢視預計算的QIIME 2物件和視覺化而不需要安裝額外的軟體(例如,QIIME 2)。
QIIME 1使用者:
在QIIME 1中,我們一般建議通過QIIME執行樣本拆分(例如,使用split_libraries.py或split_libraries_fastq.py),因為這個步驟還執行序列的質量控制。現在我們將樣本拆分和質量控制步驟分開,因此你可以使用混合多樣本序列(如我們在此所做的)或拆分後的序列開始QIIME 2分析。
拆分樣品
Demultiplexing sequences
為了混合序列進行樣本拆分,我們需要知道哪個條形碼序列與每個樣本相關聯。此資訊包含在示例元資料檔案中。你可以執行以下命令來對序列進行樣本拆分(demux emp-single命令指的是這些序列是根據地球微生物學方法新增的條形碼,並且是單端序列)。QIIME 2物件demux.qza包含樣本拆分後的序列。
qiime demux emp-single \
--i-seqs emp-single-end-sequences.qza \
--m-barcodes-file sample-metadata.tsv \
--m-barcodes-column BarcodeSequence \
--o-per-sample-sequences demux.qza
輸出結果:demux.qza
在樣本拆分之後,生成拆分結果的摘要是很有用的。這允許你確定每個樣本獲得多少序列,並且還可以獲得序列資料中每個位置處序列質量分佈的摘要。
結果統計
qiime demux summarize \
--i-data demux.qza \
--o-visualization demux.qzv
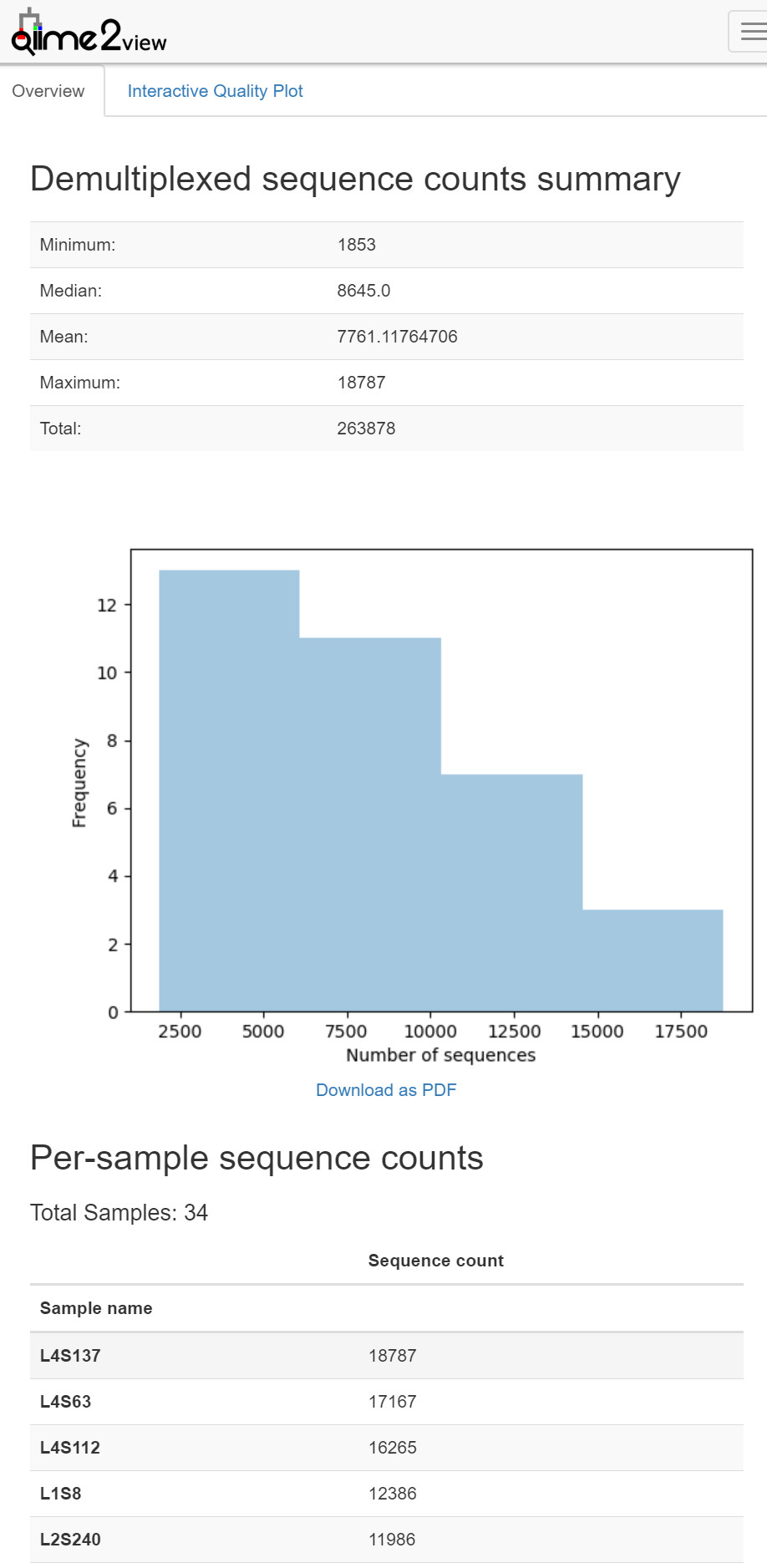
圖1. 樣本拆分結果統計結果——樣本資料量視覺化圖表。主要分為三部分:上部為摘要;中部為樣本不同資料量分佈頻率柱狀圖,可下載PDF,下部為每個樣本的測序量。上方面板還可切換至互動式質量圖Interactive Qaulity Plot
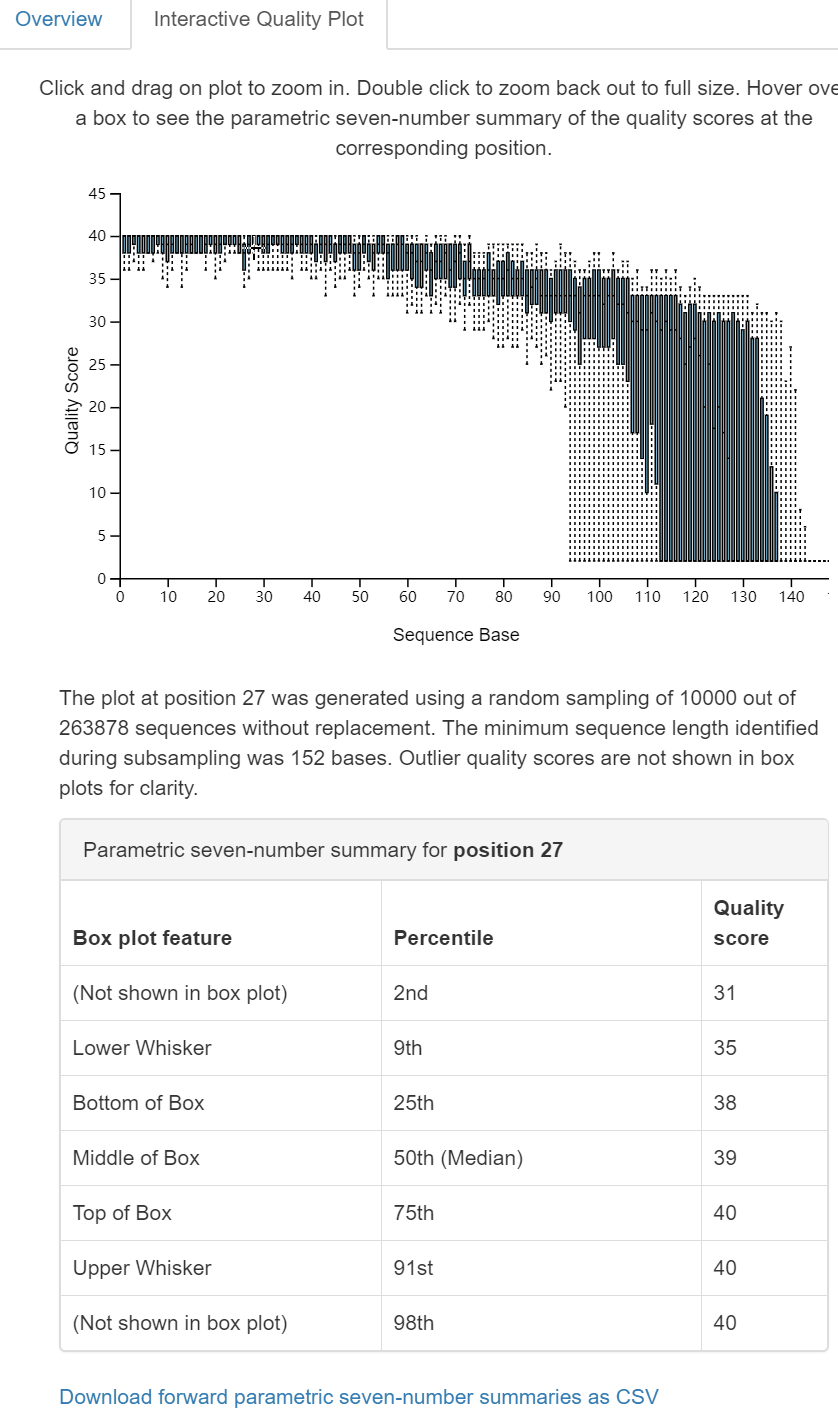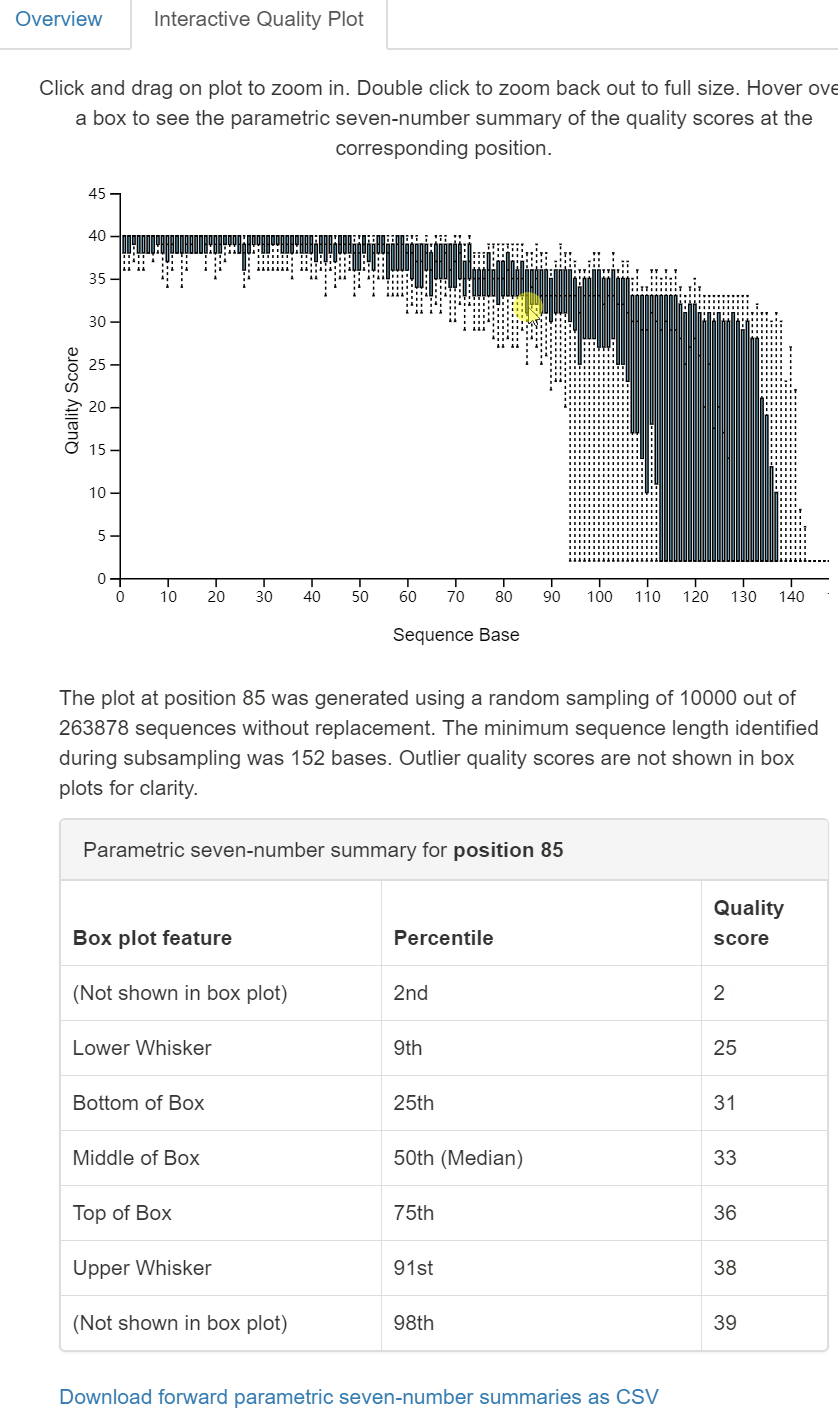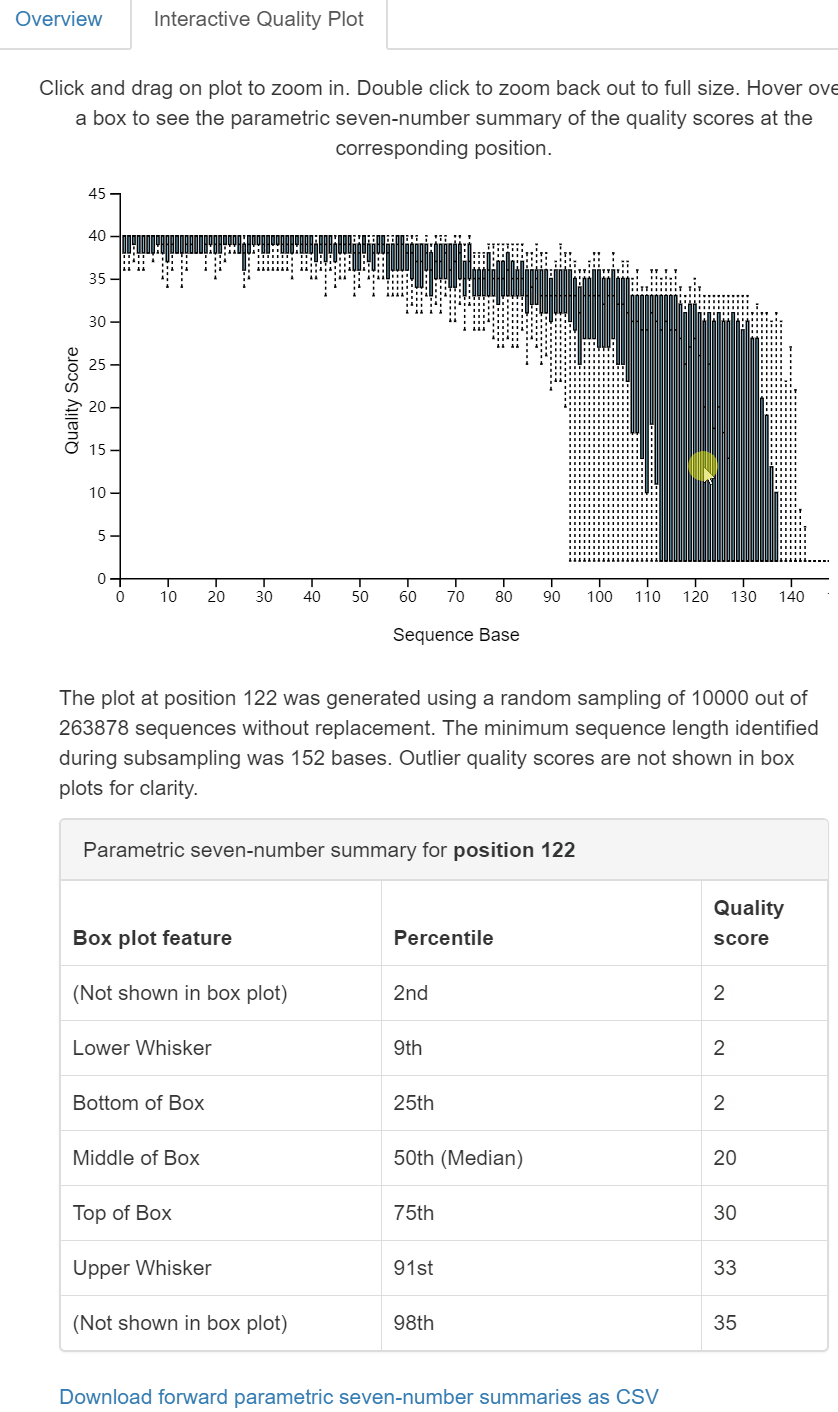
圖2. 互動式質量圖Interactive Qaulity Plot檢視頁面。同樣為三部分:上部為每個位置鹼基的質量分佈互動式箱線圖,滑鼠點選在方面(中部)文字和表格中顯示滑鼠所在位置鹼基的詳細資訊;下部為拆分樣本的長度摘要(一般無差別,略)。
所有QIIME 2視覺化物件(即,使用--o-visualization引數指定的檔案)將生成一個.qzv檔案。你可以使用qiime tools view檢視這些檔案。我們提供了用於檢視視覺化的第一個命令,但是對於本教程的其餘部分,我們將告訴你在執行視覺化程式之後檢視結果視覺化,這意味著你應該在生成的.qzv檔案上運qiime tools view。
qiime tools view demux.qzv
這條命令的顯示需要圖形介面的支援,如在有圖型介面的Linux上,但僅使用SSH登陸方式無法顯示圖形。
推薦使用 https://view.qiime2.org 網址顯示結果
可選使用[XShell+XManager]支援SSH方式的圖型介面(https://mp.weixin.qq.com/s/rbh3jynnq8E9tuhyCLtRIA)、虛擬機器或伺服器遠端桌面方式支援上面命令的圖形結果。
目前命令列方式想要檢視結果可能很多使用伺服器人員無法實現 (即依賴伺服器安裝了桌面,本地依賴XShell+XManager或其它ssh終端和圖形介面軟體)
本地檢視可解壓.qzv,目錄中的data目錄包括詳細的圖表檔案,主要關注 pdf 和 html 檔案,目錄結構如下。
── demux
└── 8743ab13-72ca-4adf-9b6c-d97e2dbe8ee3
├── checksums.md5
├── data
│ ├── data.jsonp
│ ├── demultiplex-summary.pdf
│ ├── demultiplex-summary.png
│ ├── dist
│ │ ├── bundle.js
│ │ ├── d3-license.txt
│ │ └── vendor.bundle.js
│ ├── forward-seven-number-summaries.csv
│ ├── index.html
│ ├── overview.html
│ ├── per-sample-fastq-counts.csv
│ ├── q2templateassets
│ │ ├── css
│ │ │ ├── bootstrap.min.css
│ │ │ ├── normalize.css
│ │ │ └── tab-parent.css
│ │ ├── fonts
│ │ │ ├── glyphicons-halflings-regular.eot
│ │ │ ├── glyphicons-halflings-regular.svg
│ │ │ ├── glyphicons-halflings-regular.ttf
│ │ │ ├── glyphicons-halflings-regular.woff
│ │ │ └── glyphicons-halflings-regular.woff2
│ │ ├── img
│ │ │ └── qiime2-rect-200.png
│ │ └── js
│ │ ├── bootstrap.min.js
│ │ ├── child.js
│ │ ├── jquery-3.2.0.min.js
│ │ └── parent.js
│ └── quality-plot.html
├── metadata.yaml
├── provenance
│ ├── action
│ │ └── action.yaml
│ ├── artifacts
│ │ ├── 9594ef07-c414-4658-9345-c726de100d8d
│ │ │ ├── action
│ │ │ │ └── action.yaml
│ │ │ ├── citations.bib
│ │ │ ├── metadata.yaml
│ │ │ └── VERSION
│ │ └── a7a882f3-5e4f-4b5e-8a35-6a1098d21608
│ │ ├── action
│ │ │ ├── action.yaml
│ │ │ └── barcodes.tsv
│ │ ├── citations.bib
│ │ ├── metadata.yaml
│ │ └── VERSION
│ ├── citations.bib
│ ├── metadata.yaml
│ └── VERSION
└── VERSION
qzv檔案解壓後文件詳細
序列質控和生成特徵表
Sequence quality control and feature table construction
QIIME 2外掛多種質量控制方法可選,包括DADA2、Deblur和基於基本質量分數的過濾。在本教程中,我們使用DADA2和Deblur介紹這個步驟。這些步驟是可互相替換的,因此你可以使用自己喜歡的方法。這兩種方法的結果將是一個QIIME 2物件FeatureTable[Frequency]和一個FeatureData[Sequence],Frequency物件包含資料集中每個樣本中每個唯一序列的計數(頻率),Sequence物件將FeatureTable中的特徵ID與序列對應。
譯者注:此步主要有DADA2和Deblur兩種方法可選,推薦使用DADA2,去年發表在Nature Method上,比較同類方法優於其它OTU聚類結果;相較QIIME的UPARSE聚類方法,目前DADA2方法僅去噪去嵌合,不再按相似度聚類。比上一代分析結果更精確。
注意:本節中此次存在兩種可選方法時,你將建立具有特定方法名稱的物件(例如,使用dada2去噪生成的特性表將被稱為
table-dada2.qza)。在建立這些物件之後,你將把兩個選項之一的物件重新命名為更通用的檔名(例如,table.qza)。為物件建立特定名稱,然後對其進行重新命名的過程僅允許你選擇在本步驟中使用的兩個選項中之一完成教程,而不必再次關注該選項。需要注意的是,在這個步驟或QIIME 2中的任何步驟中,你給物件或視覺化的檔案命名並不重要。
QIIME1 使用者注意:
QIIME 2物件FeatureTable[Frequency]等價於QIIME 1 OTU或BIOM表,QIIME 2物件FeatureData[Sequence]等價於QIIME 1代表序列檔案。由於DADA2和Deblur產生的“OTU”是通過對唯一序列進行分組而建立的,因此這些OTU相當於來自QIIME 1的100%相似度的OTU,通常稱為序列變體。在QIIME 2中,這些OTU比QIIME 1預設的97%相似度聚類的OTU具有更高的解析度,並且它們具有更高的質量,因為這些質量控制步驟比QIIME 1中實現更好。因此,與QIIME 1相比,可以對樣本的多樣性和分類組成進行更準確的估計。
方法1. DADA2
DADA2是用於檢測和校正(如果有可能的話)Illumina擴增序列資料的工作流程。正如在q2-dada2外掛中實現的,這個質量控制過程將過濾掉在測序資料中識別的任何phiX序列(通常存在於標記基因Illumina測序資料中,用於提高擴增子測序質量),並同時過濾嵌合序列。
譯者注:DADA2是Susan P. Holmes於2016年發表於Nature Methods的文章,截止18年12月22號Google學術統計引用483次,關於教授的工作介紹,詳見《斯坦福大學統計系教授帶你玩轉微生物組分析》;關於dada2的介紹,詳見《16S測序,不知道OTU你就out了!》;引文: Callahan, Benjamin J., Paul J. McMurdie, Michael J. Rosen, Andrew W. Han, Amy Jo A. Johnson, and Susan P. Holmes. “DADA2: high-resolution sample inference from Illumina amplicon data.” Nature methods 13, no. 7 (2016): 581.
dada2 denoise-single方法需要兩個用於質量過濾的引數:--p-trim-left m,它去除每個序列的前m個鹼基(如引物、barcode);--p-trunc-len n,它在位置n截斷每個序列。這允許使用者去除序列的低質量區域、引物等。為了確定要為這兩個引數傳遞什麼值,你應該檢視上面由qiime demux summarize生成的demux.qzv檔案中的互動質量圖選項卡。
讀者思考時間:基於上圖對拆分樣品的統計結果,如何設定
--p-trunc-len和--p-trim-left的引數值。
- –p-trim-left 擷取左端低質量序列,我們看上圖中箱線圖,左端質量都很高,無低質量區,設定為0;
- –p-trunc-len 序列擷取長度,也是為了去除右端低質量序列,我們看到大於120以後,質量下降極大,甚至中位數都下降至20以下,需要全部去除,綜合考慮決定設定為120。
單端序列去噪, 輸入樣本拆分後結果;去除左端 0 bp (–p-trim-left,有時用於切除低質量序列、barocde或引物),序列切成 120 bp 長(–p-trunc-len);生成代表序列、特徵表和去噪過程統計。
qiime dada2 denoise-single \
--i-demultiplexed-seqs demux.qza \
--p-trim-left 0 \
--p-trunc-len 120 \
--o-representative-sequences rep-seqs-dada2.qza \
--o-table table-dada2.qza \
--o-denoising-stats stats-dada2.qza
生成三個輸出檔案:
對特徵表統計進行進行視覺化
qiime metadata tabulate \
--m-input-file stats-dada2.qza \
--o-visualization stats-dada2.qzv
輸出視覺化結果:stats-dada2.qzv,特徵表統計結果,檢視 | 下載
內容為每個樣本,輸入、過濾、去噪和非嵌合的統計
sample-id input filtered denoised non-chimeric
L6S93 11270 7483 7483 7025
L6S68 9554 6169 6169 6022
我們的下游分析,將繼續使用dada2的結果,需要將它們改名方便繼續分析:
mv rep-seqs-dada2.qza rep-seqs.qza
mv table-dada2.qza table.qza
方法2. Deblur
Deblur使用序列錯誤配置檔案將錯誤的序列與從其來源的真實生物序列相關聯,從而得到高質量的序列變異資料,主要為兩個步驟。首先,應用基於質量分數的初始質量過濾過程,是Bokulich等人2013年發表的質量過濾方法。
詳者注:Deblur是本軟體作者作為通訊作者2013發表於Nature Methods的重要擴增子代表序列鑑定方法,截止18年12月22號Google學術統計引用940次,
引文:Bokulich, Nicholas A., et al. “Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing.” Nature methods 10.1 (2013): 57. 作者只將自己的方法作為了備選,分析教程中首選了dada2方法。
按測序鹼基質量過濾序列
qiime quality-filter q-score \
--i-demux demux.qza \
--o-filtered-sequences demux-filtered.qza \
--o-filter-stats demux-filter-stats.qza
輸出物件:
注意:在Deblur的論文中,作者使用了當時推薦的過濾引數。而這裡使用的引數基於最新的經驗,效果更好。
接下來,使qiime deblur denoise-16S方法應用於Deblur工作流程。此方法需要一個用於質量過濾的引數,即截斷位置n長度的序列的--p-trim-length n。通常,Deblur開發人員建議將該值設定為質量分數中位數開始下降至低質量區時的長度。在本次資料上,質量圖(在質量過濾之前)表明合理的選擇是在115至130序列位置範圍內。這是一個主觀的評估。你可能不採用該建議的一種原因是存在多個批次測序的元分析時。在這種情況的元分析中,比較所有批次的序列長度是否相同,以避免人為引入特定的偏差,全域性考慮這些是非常重要的。由於我們已經使用修剪長度為120 bp用於qiime dada2 denoise-single分析,並且由於120 bp是基於質量圖的結果,這裡我們將使用--p-trim-length 120引數。下一個命令可能需要10分鐘才能執行完成。
詳者注:deblur最大缺點就是慢,本次只分析了33個樣品,共177,092條序列。而實際研究中大專案會有成千上萬的樣本,1億-10億條序列,此步分析可能需要幾個月甚至根本無法完成,不推薦。
deblur去噪16S過程,輸入檔案為質控後的序列,設定擷取長度引數,生成結果檔案有代表序列、特徵表、樣本統計。
time qiime deblur denoise-16S \
--i-demultiplexed-seqs demux-filtered.qza \
--p-trim-length 120 \
--o-representative-sequences rep-seqs-deblur.qza \
--o-table table-deblur.qza \
--p-sample-stats \
--o-stats deblur-stats.qza
注:在我的伺服器上單執行緒運行了3m24s完成。
輸出結果:
- deblur-stats.qza: 過程統計
- table-deblur.qza: 特徵表
- rep-seqs-deblur.qza: 代表序列
注意: 本節中使用的兩個命令生成QIIME 2物件包含彙總統計資訊。為了檢視這些彙總統計資料,你可以分別使用qiime元資料表和
qiime deblur visualize-stats命令來視覺化它們。
qiime metadata tabulate \
--m-input-file demux-filter-stats.qza \
--o-visualization demux-filter-stats.qzv
qiime deblur visualize-stats \
--i-deblur-stats deblur-stats.qza \
--o-visualization deblur-stats.qzv
輸出結果:

圖3. deblur去噪和鑑定ASV處理過程統計結果
如果你想用此處結果下游分析,可以改名為下游分析的起始名稱:
mv rep-seqs-deblur.qza rep-seqs.qza
mv table-deblur.qza table.qza
記住,以上兩種方法只選擇一種即可。推薦dada2速度更快一些。有精力的條件下,可以比較一下兩種方法哪個結果更適合自己。其實每種方法都有存在的意義,而且也有適用的範圍,要在具體的專案中,結合背景知識分析哪種方法結果更好時才知道。
特徵表和特徵序列彙總
FeatureTable and FeatureData summaries
在質量篩選步驟完成之後,你將希望探索資料結果。可以使用以下兩個命令進行此操作,這兩個命令將建立資料的視覺化摘要。特性表彙總命令(feature-table summarize)將向你提供關於與每個樣品和每個特性相關聯的序列數量、這些分佈的直方圖以及一些相關的彙總統計資料的資訊。特徵表序列表格feature-table tabulate-seqs命令將提供特徵ID到序列的對映,並提供連結以針對NCBI nt資料庫輕鬆BLAST每個序列。當你想要了解關於資料集中重要的特定特性的更多資訊時,視覺化將在本教程的後續分析中非常有用。
qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv \
--m-sample-metadata-file sample-metadata.tsv
qiime feature-table tabulate-seqs \
--i-data rep-seqs.qza \
--o-visualization rep-seqs.qzv
輸出結果:

圖4. 圖中展示了特徵表的統計結果
上為摘要、中間為樣本資料量分佈和圖,下方為特徵出現頻率的統計表和圖。

圖5. 互動式檢視每組剩餘樣本量
右側還有Feature Detail進一步檢視每個特徵的頻率和在樣本中出現的次數
構建進化樹用於多樣性分析
Generate a tree for phylogenetic diversity analyses
QIIME 2支援幾種系統發育多樣性度量方法,包括Faith’s Phylogenetic Diversity、weighted和unweighted UniFrac。除了每個樣本的特徵計數(即QIIME2物件FeatureTable[Frequency])之外,這些度量還需要將特徵彼此關聯結合有根進化樹。此資訊將儲存在一個QIIME 2物件的有根系統發育物件Phylogeny[Rooted]中。為了生成系統發育樹,我們將使用q2-phylogeny外掛中的align-to-tree-mafft-fasttree工作流程。
首先,工作流程使用mafft程式執行對FeatureData[Sequence]中的序列進行多序列比對,以建立QIIME 2物件FeatureData[AlignedSequence]。接下來,流程遮蔽(mask或過濾)對齊的的高度可變區(高變區)。這些位置通常被認為會增加系統發育樹的噪聲。隨後,流程應用FastTree基於過濾後的比對結果生成系統發育樹。FastTree程式建立的是一個無根樹,因此在本節的最後一步中,應用根中點法將樹的根放置在無根樹中最長端到端距離的中點,從而形成有根樹。
qiime phylogeny align-to-tree-mafft-fasttree \
--i-sequences rep-seqs.qza \
--o-alignment aligned-rep-seqs.qza \
--o-masked-alignment masked-aligned-rep-seqs.qza \
--o-tree unrooted-tree.qza \
--o-rooted-tree rooted-tree.qza
輸出結果檔案:
- aligned-rep-seqs.qza: 多序列比對結果
- masked-aligned-rep-seqs.qza: 過濾去除高變區後的多序列比對結果
- rooted-tree.qza: 有根樹,用於多樣性分析
- unrooted-tree.qza: 無根樹
Alpha和beta多樣性分析
Alpha and beta diversity analysis
QIIME 2的多樣性分析可以通過q2-diversity外掛實現,該外掛支援計算α和β多樣性指數、並應用相關的統計檢驗以及生成互動式視覺化圖表。我們將首先應用core-metrics-phylogenetic方法,該方法將FeatureTable[Frequency](特徵表[頻率])抽平到使用者指定的測序深度,計算常用幾種α和β多樣性指數,並使用Emperor為每個β多樣性指數生成主座標分析(PCoA)圖。預設情況下計算的方法有:
劃重點:理解下面4種alpha和beta多樣性指數的所代表的生物學意義至關重要。
- α多樣性
- 夏農(Shannon’s)多樣性指數(群落豐富度的定量度量,即包括豐富度
richness和均勻度evenness兩個層面) - Observed OTUs(群落豐富度的定性度量,只包括豐富度)
- Faith’s系統發育多樣性(包含特徵之間的系統發育關係的群落豐富度的定性度量)
- 均勻度(或 Pielou’s均勻度;群落均勻度的度量)
- 夏農(Shannon’s)多樣性指數(群落豐富度的定量度量,即包括豐富度
- β多樣性
- Jaccard距離(群落差異的定性度量,即只考慮種類,不考慮丰度)
- Bray-Curtis距離(群落差異的定量度量)
- 非加權UniFrac距離(包含特徵之間的系統發育關係的群落差異定性度量)
- 加權UniFrac距離(包含特徵之間的系統發育關係的群落差異定量度量)
需要提供給這個指令碼的一個重要引數是--p-sampling-depth,它是指定重取樣(即稀疏/稀釋)深度。因為大多數多樣指數對不同樣本的不同測序深度敏感,所以這個指令碼將隨機地將每個樣本的測序量重新取樣至該引數提供的值。例如,如果提供--p-sampling-depth 500,則此步驟將對每個樣本中的計數進行無放回抽樣,從而使得結果表中的每個樣本的總計數為500。如果任何樣本的總計數小於該值,那麼這些樣本將從多樣性分析中刪除。選擇這個值很棘手。我們建議你通過檢視上面建立的表table.qzv檔案中呈現的資訊並選擇一個儘可能高的值(因此每個樣本保留更多的序列)同時儘可能少地排除樣本來進行選擇。
讀者思考時間:
檢視QIIME 2的table.qzv物件,尤其是互動式視覺化表格。對於取樣深度p,應該選擇什麼值呢?根據這個選擇,分析中多少個樣本將被排除?在core-metrics-phylogenetic命令中,你將分析的總序列是多少條呢?
譯者注:下面多樣性分析,需要基於標準化的特徵表,標準化採用無放回重抽樣至序列一致,如何設計樣品重抽樣深度引數
--p-sampling-depth呢?
如是資料量都很大,選最小的即可。如果有個別數據量非常小,去除最小值再選最小值。比如此分析最小值為917,我們選擇1109深度重抽樣,即保留了大部分樣品用於分析,又去除了資料量過低的異常值。
注:本示例為近10年前測序技術的通量水平,現在看來資料量很小。目錄一般採用HiSeq PE250測序,資料量都非常大,通常可以採用3萬或5萬的標準篩選,仍可保留90%以上樣本。過低或過高一般結果也會波動較大,不建議放在一起分析。
計算核心多樣性
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 1109 \
--m-metadata-file sample-metadata.tsv \
--output-dir core-metrics-results
輸出物件(資料檔案):
- core-metrics-results/faith_pd_vector.qza: Alpha多樣性考慮進化的faith指數
- core-metrics-results/unweighted_unifrac_distance_matrix.qza: 無權重unifrac距離矩陣
- core-metrics-results/bray_curtis_pcoa_results.qza: 基於Bray-Curtis距離PCoA的結果
- core-metrics-results/shannon_vector.qza: Alpha多樣性夏農指數
- core-metrics-results/rarefied_table.qza: 抽樣後的特徵表
- core-metrics-results/weighted_unifrac_distance_matrix.qza: 有權重的unifrac距離矩陣
- core-metrics-results/jaccard_pcoa_results.qza: jaccard距離PCoA結果
- core-metrics-results/observed_otus_vector.qza: Alpha多樣性observed otus指數
- core-metrics-results/weighted_unifrac_pcoa_results.qza: 基於有權重的unifrac距離的PCoA結果
- core-metrics-results/jaccard_distance_matrix.qza: jaccard距離矩陣
- core-metrics-results/evenness_vector.qza: Alpha多樣性均勻度指數
- core-metrics-results/bray_curtis_distance_matrix.qza: Bray-Curtis距離矩陣
- core-metrics-results/unweighted_unifrac_pcoa_results.qza: 無權重的unifrac距離的PCoA結果
輸出物件(視覺化結果):
- core-metrics-results/unweighted_unifrac_emperor.qzv: 無權重的unifrac距離PCoA結果採用emperor視覺化。 檢視 | 下載
- core-metrics-results/jaccard_emperor.qzv: jaccard距離PCoA結果採用emperor視覺化。檢視 | 下載
- core-metrics-results/bray_curtis_emperor.qzv: Bray-Curtis距離PCoA結果採用emperor視覺化。檢視 | 下載
- core-metrics-results/weighted_unifrac_emperor.qzv: 有權重的unifrac距離PCoA結果採用emperor視覺化。檢視 | 下載
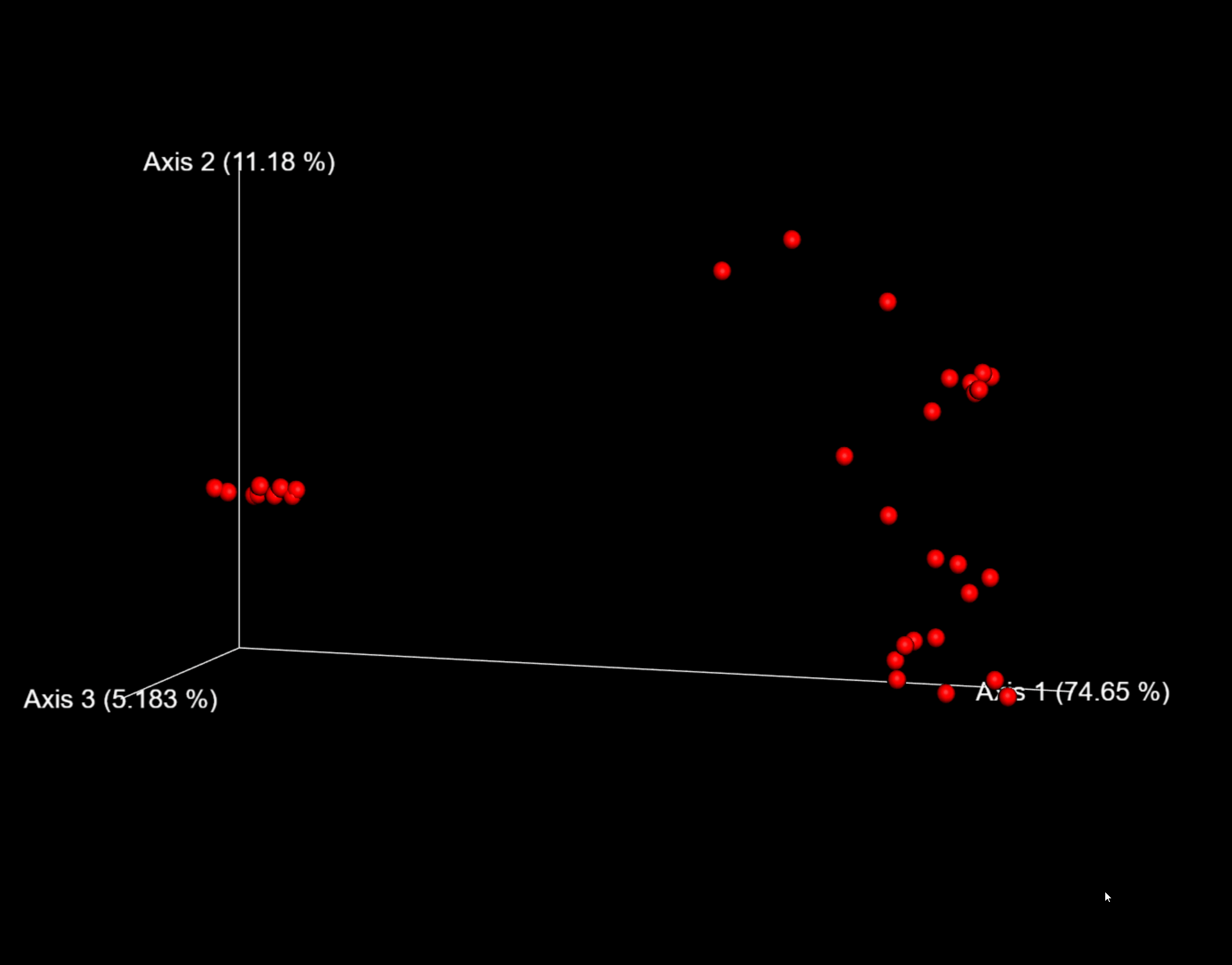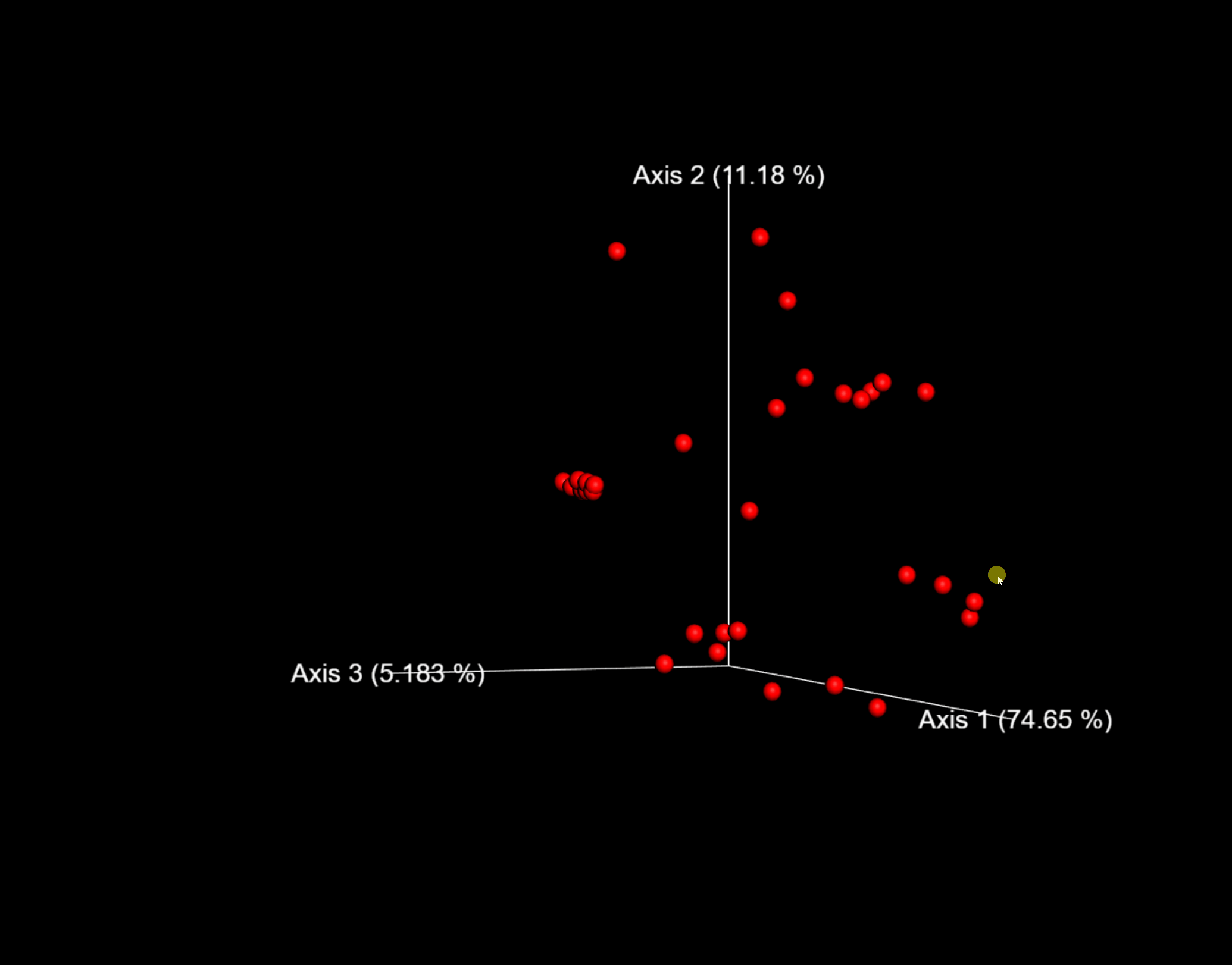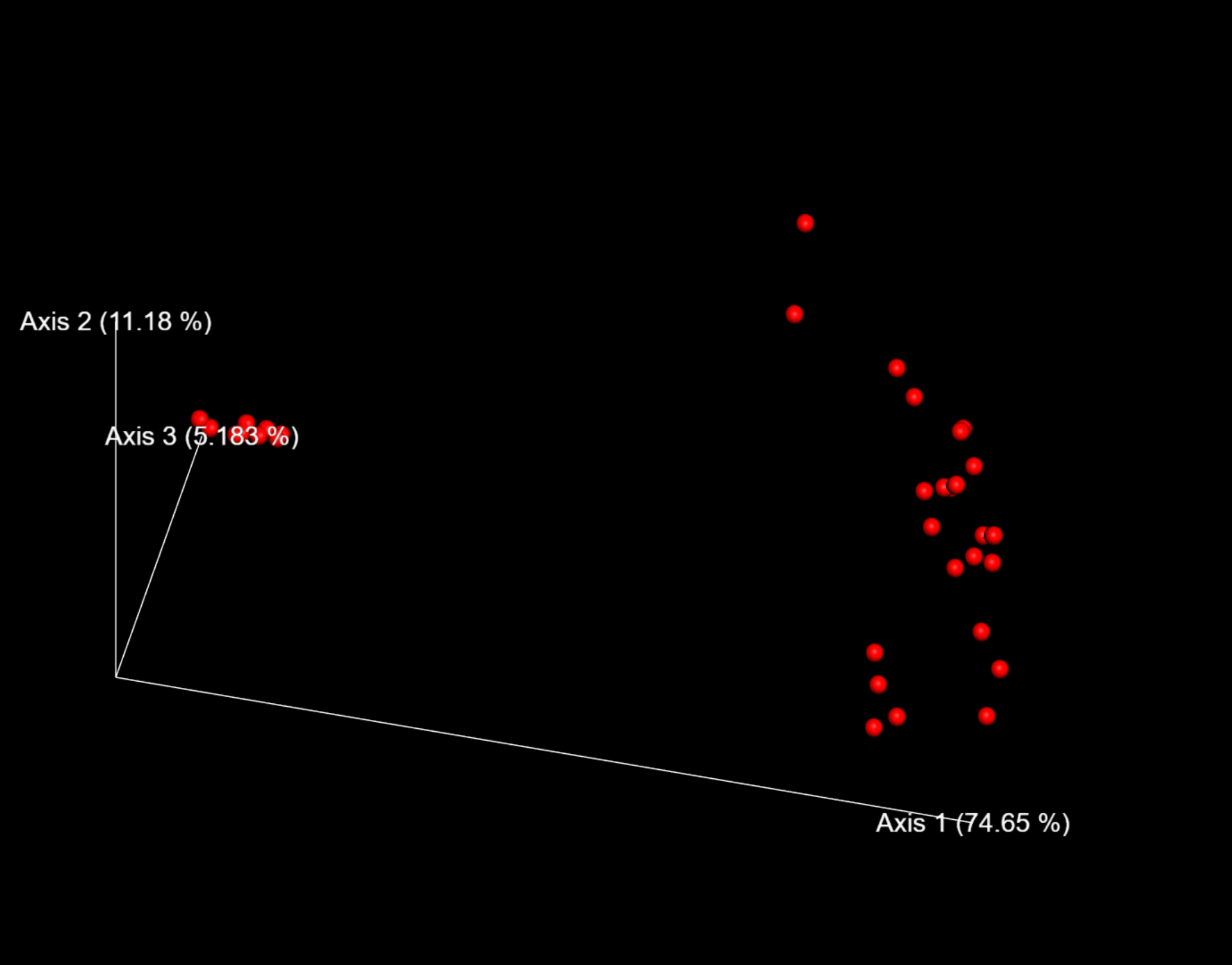
圖6. 以weighted_unifrac距離的PCoA結果互動式視覺化為例,可用滑鼠託動空間檢視每個樣本的分佈位置。
這裡,我們將--p-sampling-depth引數設定為1109。這個值是根據L3S341樣本中的序列數量來選擇的,因為它與接下來幾個序列計數較高的樣本中的序列數量接近,並且因為它僅比序列較少的一個樣本中的序列數量高。這將允許我們保留大部分樣品。具有較少序列的一個樣本將從core-metrics-phylogenetic分析和任何使用這些結果的任何下游分析中刪除。
注意:根據DADA2特徵表彙總選擇1109的取樣深度。如果使用的是Deblur特性表而不是DADA2特性表,則可能需要選擇不同的取樣深度。應用上一段的邏輯來幫助你選擇合理的取樣深度。
注意:在許多Illumina測序結果中,你將觀察到一些序列計數非常低的例子。你通常希望通過在此階段取樣深度選擇更大的值來從分析中剔除它們。
在計算多樣性度量之後,我們可以開始在樣本元資料的分組資訊或屬性值背景下探索樣本的微生物組成差異。此資訊存在於先前下載的示例元資料檔案中。
我們將首先測試分類元資料列和alpha多樣性資料之間的關係。我們將在這裡為Faith系統發育多樣性(群體豐富度的度量)和均勻度進行視覺化操作。
Alpha多樣性組間顯著性分析和視覺化
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/faith_pd_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/faith-pd-group-significance.qzv
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/evenness_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/evenness-group-significance.qzv
輸出視覺化結果:
- core-metrics-results/faith-pd-group-significance.qzv。檢視 | 下載
- core-metrics-results/evenness-group-significance.qzv。檢視 | 下載
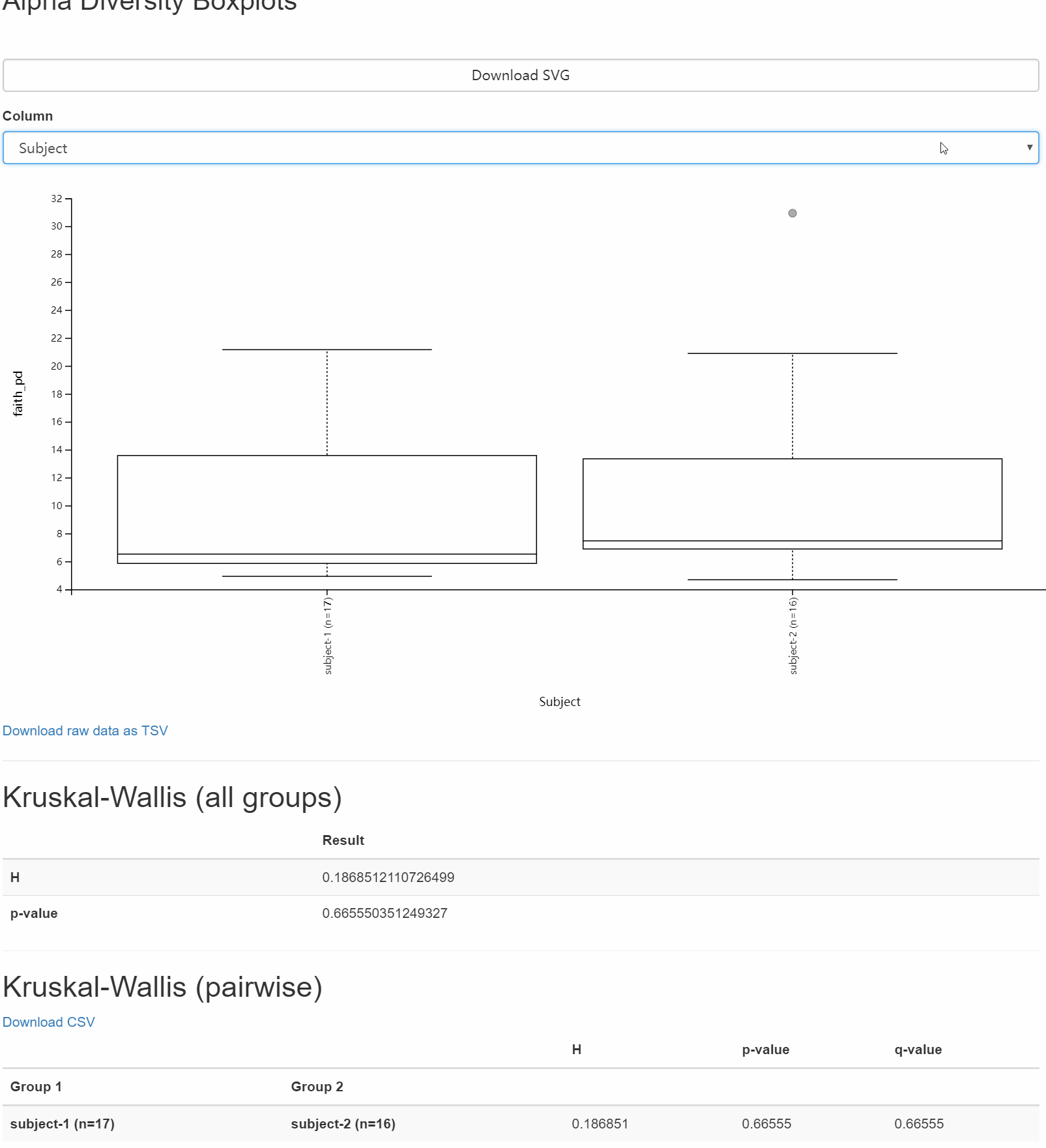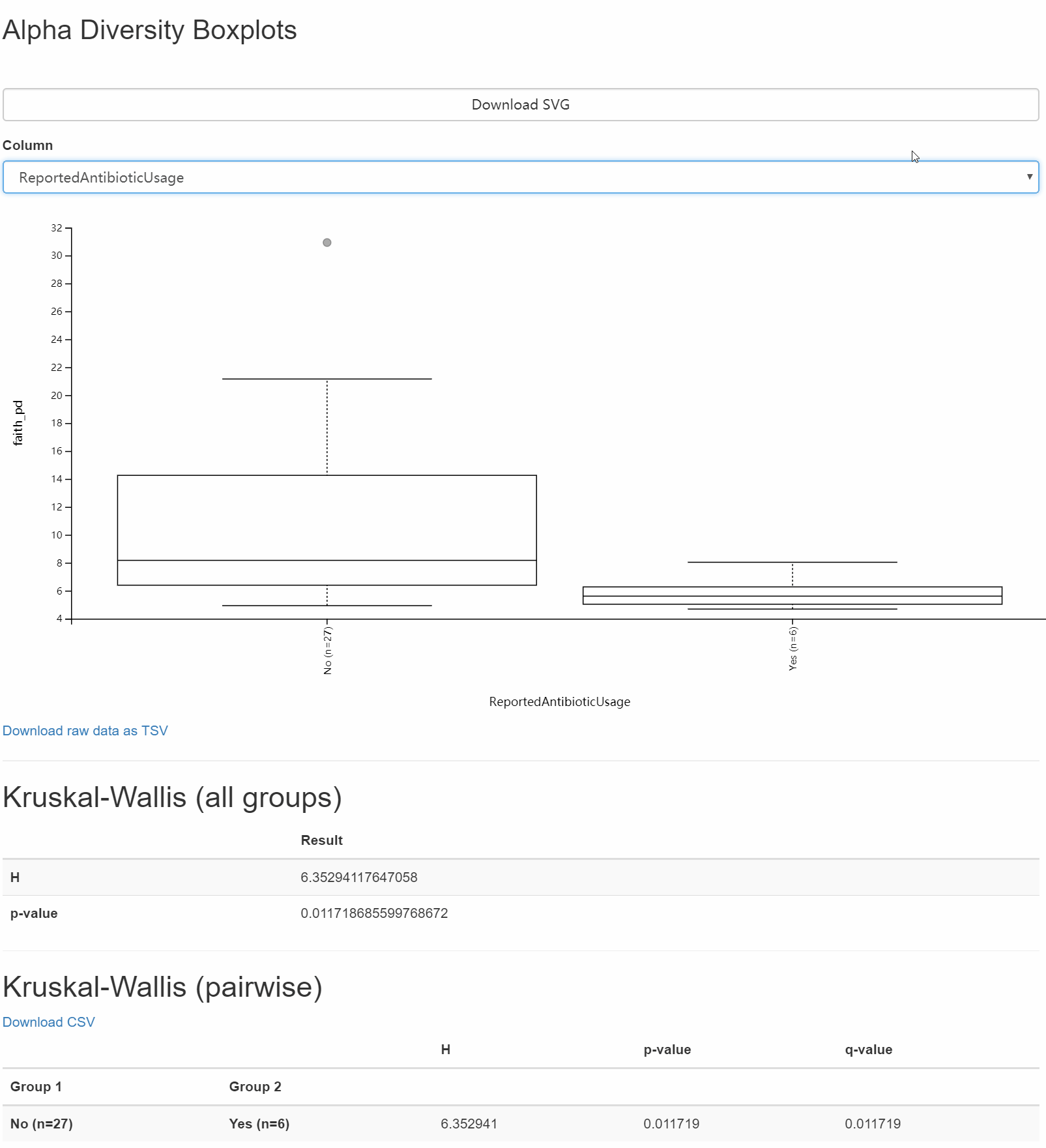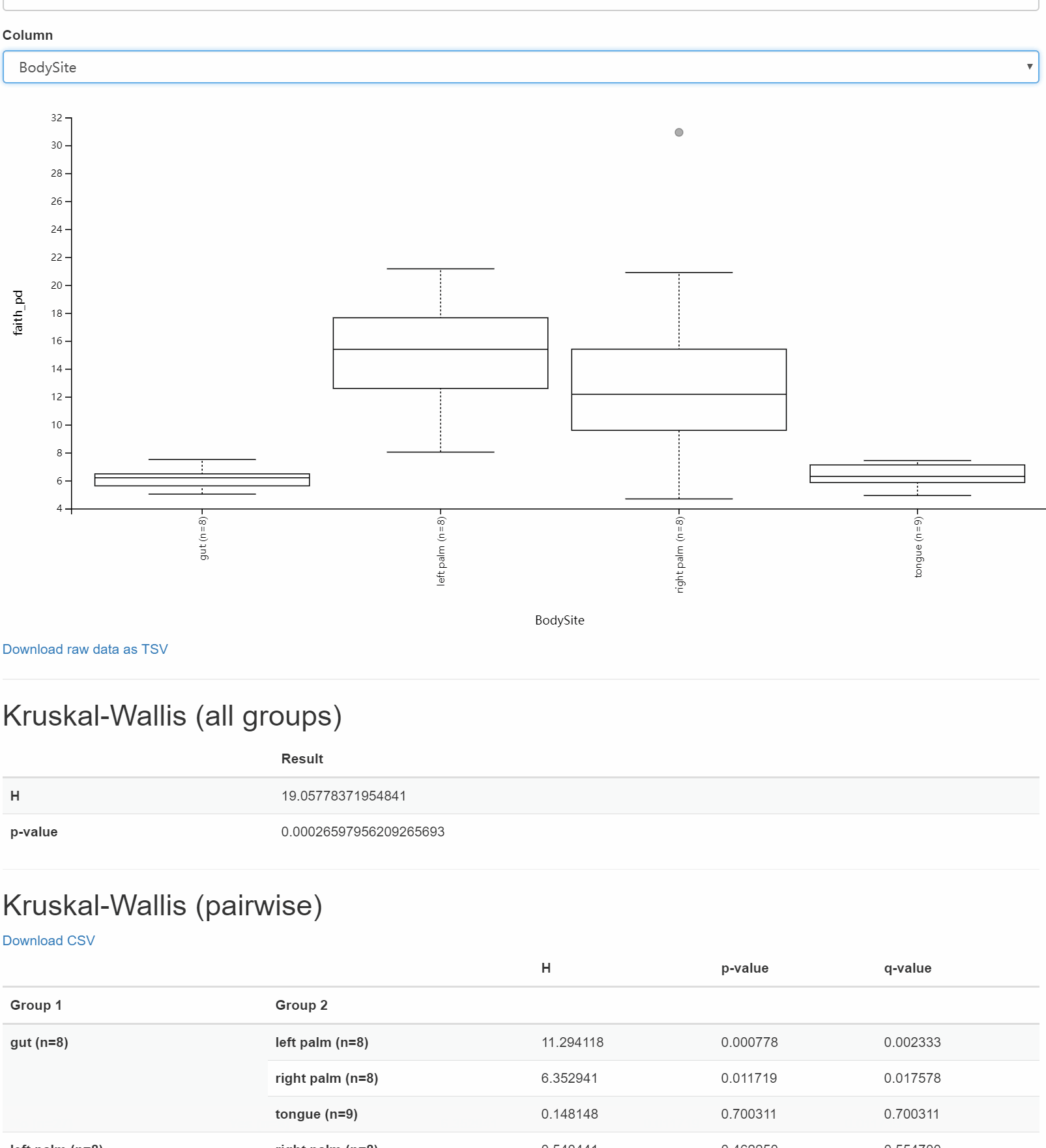
圖7. 以faith-pd為例將互探索不同元資料條件下組間差異,可用滑鼠選擇不同元資料的列名,切換分組方式,探索對應的生物學意義。
問題:哪些分類樣本元資料列與微生物群落豐富度的差異密切相關?這些差異在統計學上有顯著性嗎?
問題:哪些分類樣本元資料列與微生物群落均勻性的差異密切相關?這些差異在統計學上有顯著性嗎?
讀者思考時間:實驗設計中的那一種分組方法,與微生物群體的豐富度差異相關,這些差異顯著嗎?
解答:圖中可按Catalogy選擇分類方法,檢視不同分組下箱線圖間的分佈與差別。圖形下面的表格,詳細詳述組間比較的顯著性和假陽性率統計。
結果我們會看到本實驗設計的分組方式有Bodysite, Subject, ReportAntibioticUse,只有身體位置各組間差異明顯,且下面統計結果也存在很多組間的顯著性差異。
在這個資料集中,沒有連續的樣本元資料列(例如,DaysSinceExperimentStart)與α多樣性相關聯,所以我們這裡不測試這類關聯。如果你有興趣執行這類測試(對於這個資料集或其他資料集),可以使用qiime diversity alpha-correlation命令。
接下來,我們將使用PERMANOVA方法(首先在Anderson 2001年的文章中描述)beta-group-significance分析分類元資料背景下的樣本組合。以下命令將測試一組樣本之間的距離,是否比來自其他組(例如,舌頭、左手掌和右手掌)的樣本彼此更相似,例如來自同一身體部位(例如腸)的樣本。如果你用這個命令的--p-pairwise引數,它將執行成對檢驗,結果將允許我們確定哪對特定組(例如,舌頭和腸)彼此不同是否顯著不同。這個命令執行起來可能很慢,尤其是當使用--p-pairwise引數,因為它是基於置換檢驗的。因此,我們將在元資料的特定列上執行該命令,而不是在其適用的所有元資料列上執行該命令。這裡,我們將使用兩個示例元資料列將此應用到未加權的UniFrac距離,如下所示。
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-column BodySite \
--o-visualization core-metrics-results/unweighted-unifrac-body-site-significance.qzv \
--p-pairwise
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-column Subject \
--o-visualization core-metrics-results/unweighted-unifrac-subject-group-significance.qzv \
--p-pairwise
輸出視覺化結果:
core-metrics-results/unweighted-unifrac-body-site-significance.qzv: 檢視 | 下載core-metrics-results/unweighted-unifrac-subject-group-significance.qzv: 檢視 | 下載

圖8. 不同部分組內和組間差異顯著性分析箱線圖+統計表
問題
受試者之間的關聯和微生物組成的差異在統計學上是否顯著?身體部位呢?哪些特定的身體部位對彼此有顯著的不同?
同樣,我們對於這個資料集所擁有的連續樣本元資料中沒有一個與樣本組成相關,因此這裡我們不會測試這些關聯。如果你對執行這些測試感興趣,那麼可以使用qiime metadata distance-matrix、qiime diversity mantel和qiime diversity bioenv命令組合使用。
最後,排序是在樣本元資料分組間探索微生物群落組成差異的流行方法。我們可以使用Emperor工具在示例元資料背景下探索主座標(PCoA)繪圖。雖然我們的core-metrics-phylogenetic命令已經生成了一些Emperor圖,但我們希望傳遞一個可選的引數--p-custom-axes,這對於探索時間序列資料非常有用。用於core-metrics-phylogeny的PCoA結果也是可用的,這使得很容易與Emperor生成新的視覺化。我們將為未加權的UniFrac和Bray-Curtis的PCoA結果生成Emperor圖,以便所得到的圖將包含主座標1、主座標2和實驗開始以來的天數(days since the experiment start)的軸。我們將使用最後一個軸來探索這些樣本是如何隨時間變化的。
qiime emperor plot \
--i-pcoa core-metrics-results/unweighted_unifrac_pcoa_results.qza \
--m-metadata-file sample-metadata.tsv \
--p-custom-axes DaysSinceExperimentStart \
--o-visualization core-metrics-results/unweighted-unifrac-emperor-DaysSinceExperimentStart.qzv
qiime emperor plot \
--i-pcoa core-metrics-results/bray_curtis_pcoa_results.qza \
--m-metadata-file sample-metadata.tsv \
--p-custom-axes DaysSinceExperimentStart \
--o-visualization core-metrics-results/bray-curtis-emperor-DaysSinceExperimentStart.qzv
輸出視覺化:
- core-metrics-results/unweighted-unifrac-emperor-DaysSinceExperimentStart.qzv: 檢視 | 下載
- core-metrics-results/bray-curtis-emperor-DaysSinceExperimentStart.qzv: 檢視 | 下載

圖9. 探索樣本在第1/2主軸和時間上的分佈,調整右側著色方式和顏色方案可方便觀察研究的分類或時間序列結果。
問題:Emperor圖是否支援我們在這裡執行的其他β多樣性分析?(提示:對不同實驗元資料進行點著色。)
問題:在未加權的UniFrac和Bray-Curtis PCoA圖中,你觀察到了哪些差異?
Alpha稀釋取線
Alpha rarefaction plotting
在本節中,我們將使用qiime diversity alpha-rarefaction視覺化器來探索α多樣性與取樣深度的關係。該視覺化器在多個取樣深度處計算一個或多個α多樣性指數,範圍介於1(可選地--p-min-depth控制)和作為--p-max-depth提供值之間。在每個取樣深度,將生成10個抽樣表,並對錶中的所有樣本計算alpha多樣性指數計算。迭代次數(在每個取樣深度計算的稀疏表)可以通過--p-iterations來控制。在每個取樣深度,將為每個樣本繪製平均多關性值,如果提供樣本元資料--m-metadata-file引數,則可以基於元資料對樣本進行分組。
qiime diversity alpha-rarefaction \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \
--p-max-depth 4000 \
--m-metadata-file sample-metadata.tsv \
--o-visualization alpha-rarefaction.qzv
輸出視覺化:

圖10. 檢視按body site分組下observed otus的稀疏箱線圖,注意觀察圖中變化以及下面對應樣本資料的圖。
視覺化將有兩個圖。頂部圖是α稀疏圖,主要用於確定樣品的丰度是否已被完全觀察或測序。如果圖中的線條在沿x軸的某個取樣深度處看起來“平坦”(即,接近於零的斜率),這表明收集超過該取樣深度的附加序列不太可能導致對附加特徵的觀察。如果繪圖中的線條沒有變平,這可能是因為尚未充分觀察樣本的豐富度(因為收集的序列太少),或者它可能是在資料中仍然存在許多測序錯誤(這被誤認為是新的多樣性)的指標。
當通過元資料對樣本進行分組時,此視覺化中的底部繪圖非常重要。它說明了當特徵表被細化到每個取樣深度時,每個組中剩餘的樣本數量。如果給定的取樣深度d大於樣本s的總頻率(即,針對樣本s獲得的序列數),則不可能計算取樣深度d下樣本s的多樣性。在頂部繪圖將不可靠,因為它將計算基於相對少的樣本。因此,當通過元資料對樣本進行分組時,必須檢視底部圖表,以確定頂部圖表中顯示的資料是否可靠的。
注意:提供的
--p-max-depth引數的值應該通過檢視上面建立的table.qzv檔案中呈現的“每個樣本的測序量”資訊來確定。一般來說,選擇一個在中位數附近的值似乎很好用。如果得到的稀疏圖中的線看起來沒有變平,那麼你可能希望增加該值。如果由於大於最大采樣深度而丟失了許多樣本,則減少該值。
問題1:當通過“BodySite”列資訊對樣本進行分組並檢視“observed_otus”指數的α稀疏圖時,哪些身體部位顯示出足夠的多樣性覆蓋(即稀疏曲線趨於平緩)?在這些身體部位似乎存在多少序列變異?
問題2:當通過“BodySite”對樣本進行分組並檢視“observed_otus”指數的α稀疏圖時,“右手掌(right palm)”樣本的線看起來在40左右變平,但隨後跳到大約140。你認為這裡發生了什麼?(提示:一定要檢視頂部和底部的細節。)
譯者注問題2答案:左手掌的多樣性從突然40跳至140,而對應的樣本量從9個下降為3個(由於測序深度不足)。僅有3次生物學重複樣本量太少,偶然性太大,導致的結果波動可信度不高。問題1很簡單,自己看圖吧可以想出答案。
物種組成分析
在這一節中,我們將開始探索樣本的物種組成,並將其與樣本元資料再次組合。這個過程的第一步是為FeatureData[Sequence]的序列進行物種註釋。我們將使用經過Naive Bayes分類器預訓練的,並由q2-feature-classifier外掛來完成這項工作。這個分類器是在Greengenes 13_8 99% OTU上訓練的,其中序列被修剪到僅包括來自16S區域的250個鹼基,該16S區域在該分析中採用V4區域的515F/806R引物擴增並測序。我們將把這個分類器應用到序列中,並且可以生成從序列到物種註釋結果關聯的視覺化。
注意:物種分類器根據你特定的樣品製備和測序引數進行訓練時表現最好,包括用於擴增的引物和測序序列的長度。因此,一般來說,你應該按照使用
q2-feature-classifier的訓練特徵分類器的說明來訓練自己的物種分類器。我們在資料資源頁面上提供了一些通用的分類器,包括基於Silva的16S分類器,不過將來我們可能會停止提供這些分類器,而讓使用者訓練他們自己的分類器,這將與他們的序列資料最相關。
下載物種註釋資料庫製作的分類器:無法下載記得後臺回覆"qiime2"獲得備用下載連結
wget \
-O "gg-13-8-99-515-806-nb-classifier.qza" \
"https://data.qiime2.org/2018.11/common/gg-13-8-99-515-806-nb-classifier.qza"
物種註釋和視覺化
qiime feature-classifier classify-sklearn \
--i-classifier gg-13-8-99-515-806-nb-classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
輸出結果:
- taxonomy.qza: 物種註釋結果
- gg-13-8-99-515-806-nb-classifier.qza: 分類器的訓練結果
視覺化結果:
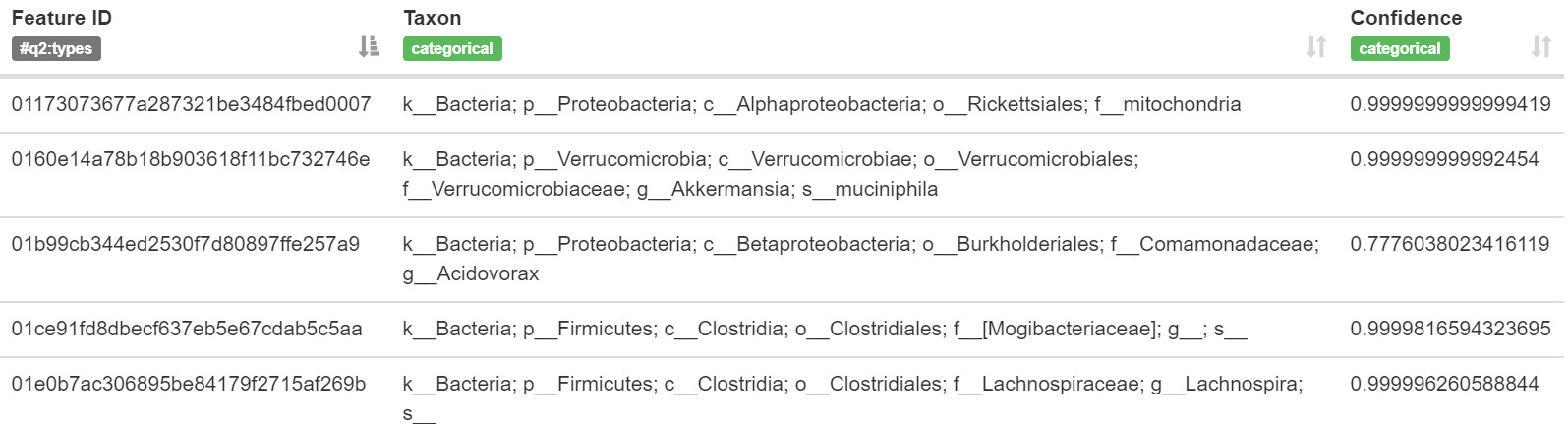
圖11. ID對應的物種分類和置信度
問題:回想一下,
rep-seqs.qzv視覺化允許你輕鬆地對NCBI nt資料庫BLAST每個特性的序列。使用此處建立的視覺化和taxonomy.qzv視覺化,將幾個特性物種分配與最佳BLAST命中的分類進行比較,結果有多相似?如果它們不同,它們在什麼分類學層次上開始不同(例如,物種、屬、科…)?
接下來,我們可以用互動式條形圖檢視樣本的分類組成。使用以下命令繪圖成堆疊柱狀圖,然後開啟視覺化。
qiime taxa barplot \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization taxa-bar-plots.qzv
結果:
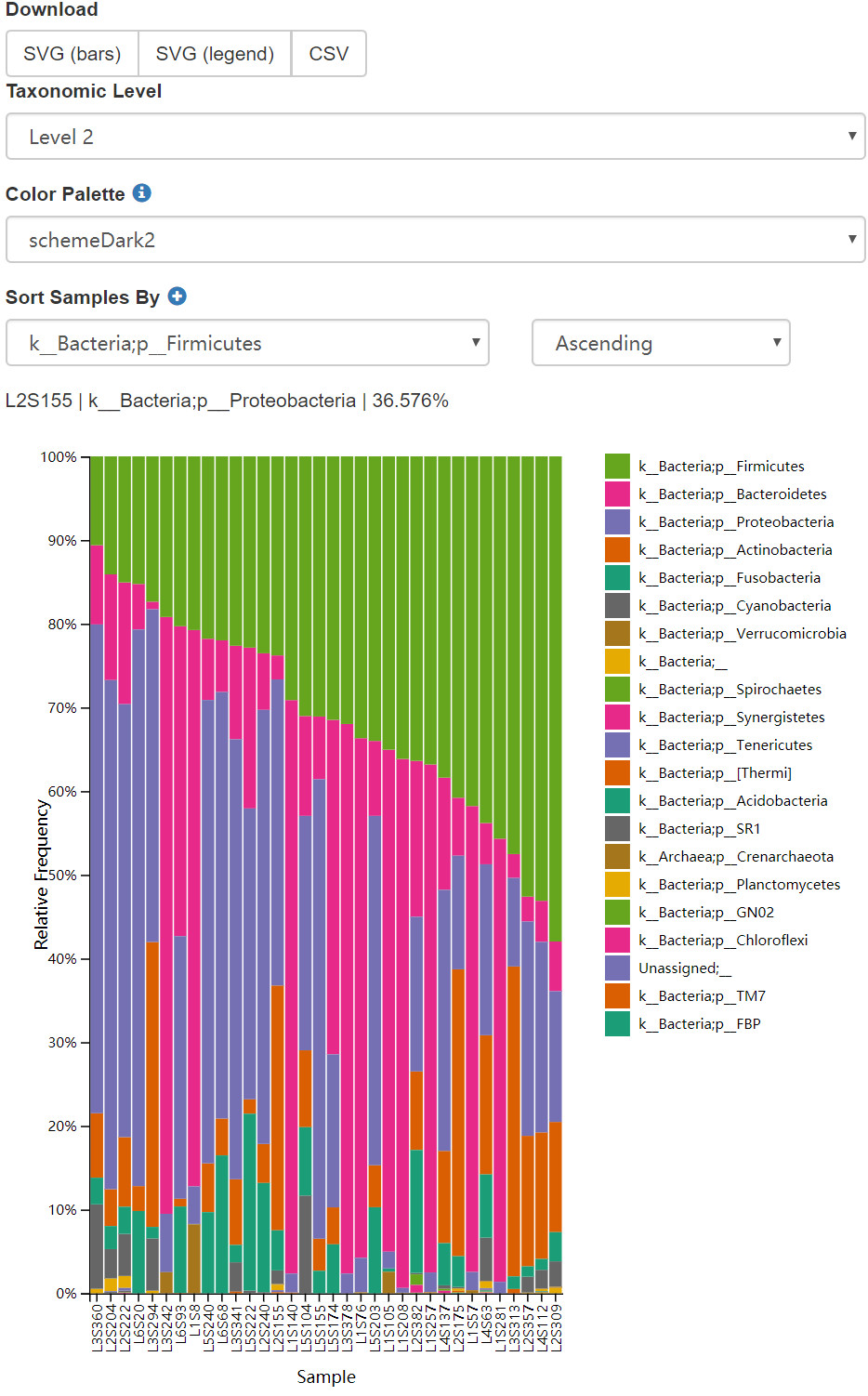
圖12. 門水平樣本堆疊柱狀圖、按Firmicutes排序。可切換不同分類級別、選擇10餘種本色方案; 切換排序各類和方向。
問題:在物種註釋第二級視覺化樣本(在本分析中對應於門級別),然後按
BodySite、Subject、然後按DaysSinceExperimentStart對樣本進行排序。在BodySite中不同部位都有哪些優勢門類?在DaysSinceExperimentStart0和後面的時間點之間,你是否觀察到兩個組之間的任何一致的變化規律?
使用ANCOM差異丰度分析
Differential abundance testing with ANCOM
ANCOM可用於識別不同樣本組中丰度差異的特徵。與任何生物資訊學方法一樣,在使用ANCOM之前,你應該瞭解ANCOM的假設和侷限性。我們建議在使用這種方法之前先回顧一下ANCOM的論文 https://www.ncbi.nlm.nih.gov/pubmed/26028277。
注意:差異丰度檢驗在微生物學分析中是一個熱門的研究領域。有兩個QIIME 2外掛可用:
q2-gneiss和q2-composition。本節使用q2-composition,但是如果你想了解更多,還有一個教程在另外的資料集上使用q2-gneiss,在後面有詳細介紹。
ANCOM是在q2-composition外掛中實現的。ANCOM假設很少(小於約25%)的特徵在組之間改變。如果你期望在組之間有更多的特性正在改變,那麼就不應該使用ANCOM,因為它更容易出錯(I類和II類錯誤都有可能增加)。因為我們預期身體部位的許多特徵都會發生變化,所以在本教程中,我們將過濾完整的特徵表後只包含腸道樣本。然後,我們將應用ANCOM來確定哪種(如果有的話)序列變體在我們兩個受試者的腸道樣本中丰度存在差異。
我們將首先建立一個只包含腸道樣本的特徵表。(要了解關於篩選的更多資訊,請參閱資料篩選教程。)
qiime feature-table filter-samples \
--i-table table.qza \
--m-metadata-file sample-metadata.tsv \
--p-where "BodySite='gut'" \
--o-filtered-table gut-table.qza
輸出物件:
- gut-table.qza:只包含腸道樣本的OTU表
ANCOM基於每個樣本的特徵頻率對FeatureTable[Composition]進行操作,但是不能容忍零。為了構建組成物件,必須提供一個新增偽計數(一種遺失值插補方法)的FeatureTable[Frequency]物件,這將產生FeatureTable[Composition]物件。
qiime composition add-pseudocount \
--i-table gut-table.qza \
--o-composition-table comp-gut-table.qza
輸出結果:
- comp-gut-table.qza: 組成型特徵表
接下來可用ANCON對兩組的特徵進行丰度差異的比較了。
qiime composition ancom \
--i-table comp-gut-table.qza \
--m-metadata-file sample-metadata.tsv \
--m-metadata-column Subject \
--o-visualization ancom-Subject.qzv
輸出結果:

圖13. 互動火山圖展示組間差異特徵。滑鼠懸停在特徵點上,可顯示特徵名稱和對應的具體座標。下面有每個顯著差異特徵的統計結果,以及組內分位數表格。
問題:哪個ASV在分組間差異很大?每個序列變體在哪個分組中更豐富?這些序列變體的分類是什麼?(要回答最後一個問題,你需要參考本教程中生成的另一個視覺化。)
我們也經常對在特定的分類學層次上執行差異丰度檢驗。為此,我們可以在感興趣的分類級別上摺疊FeatureTab