
RNA提取和建庫流程對mRNA-Seq的影響
RNA提取和建庫流程對mRNA-Seq的影響
目前RNA-Seq是挖掘不同生長時期及不同脅迫條件下、不同組織細胞中其差異表達基因通常所採用的研究方法,同時還可以鑑定獲得新的轉錄本資訊以及不同的可變剪下事件,因而RNA-Seq目前應用很廣泛。結合不同的RNA提取方法及文庫構建流程對RNA-Seq獲得的測序資料產生不同的影響。
1.關於總RNA提取
關於RNA提取對於大家最為熟悉的是Trizol-based的RNA提取方法,也有結合試劑盒來進行提取的。在提取總RNA的過程中通常會引入影響後續PCR酶促反應的抑制劑等,這些抑制劑如不正確去除的話,會對後續的反轉錄、末端修復、加A以及接頭連線和PCR擴增等產生影響,如阻礙聚合酶的聚合、影響聚合酶的活性甚至降解聚合酶等,從而對最終獲得的測序資料造成影響。
樣本中常見的抑制劑包含由樣本中本身就帶有的和在實驗操作過程中帶入的,樣本中本身包含的抑制劑如血液中的血紅蛋白,植物樣本中的腐殖酸、黃腐酸等;在實驗過程中帶入的抑制劑如EDTA、肝素、氯酚仿等。不同樣本中可能引入的抑制劑或其他汙染物會不一樣,詳見
如果樣本中存在這些抑制劑等汙染物質的話,需結合試劑盒進一步進行純化,比如過柱子過濾的試劑盒等,達到總RNA理想標準方可開展後續實驗。
總RNA提取結果檢測標準:
總RNA溶解環境:ph7.5-8.0;
結合Qubit or Pico/RiboGreen/Agilent 2100進行檢測;
Substance Absorbance (nm) 260/280 Ratio Values 260/230 Ratio Values
Pure DNA 280 nm ~1.8 2.0–2.2
Pure RNA 280 nm ~2.0 2.0–2.2
EDTA, Carbohydrates, Phenol 230 nm < 1.5 < 2.0
Guanidine HCL 230 nm < 1.5 < 2.0
2.關於去除rRNA
考慮到總RNA中含有大量的rRNA序列,大約是在80%-90%的序列是rRNA,因而會結合不同的方法來去除總RNA中的rRNA。真核生物種常規的去除rRNA的方法是通過oligo(dT)富集帶有polyA尾的mRNA來實現的,但是這種方法針對不含有polyA尾的轉錄本序列以及存在部分降解的總RNA樣本,所以這種方法針對FF(Formalin-Fixed)樣本和FFPE(Paraffin-Embedded)石蠟包埋樣本是不適用的,否則對獲得樣本中最全面的轉錄本資訊會產生顯著影響。
針對於FF和FFPE樣本以及原核生物的總RNA中去除rRNA,則需結合RiboZero、RiboMinus等是結合來開展去除,其實針對rRNA序列進行雜交捕獲去除的原理來去除的。針對FFPE樣本還有結合雙鏈特異性核酸酶構建文庫來降低後續測序資料中的rRNA序列比例的。
常見去除rRNA方法:
a. rRNA消減雜交法:相應的試劑盒有MICROBExpress bacterial mRNA enrichment kit (Ambion),RiboMinus bacteria transcriptome isolation kit (Invitrogen) 和Ribo-Zero rRNA removal kit (Epicentre);
b. 5′單核苷酸依賴的外切酶處理法:相應的試劑盒主要有mRNA-ONLY prokaryotic mRNA isolation kit (Epicentre);
c. 選擇性引物擴增法:相應試劑盒主要有Ovation prokaryotic RNA-seq system (NuGEN);
d. 依賴於雙鏈特異核酸酶的cDNA均一化法:相應的試劑盒主要有trimmer-direct cDNA normalization kit(Evrogen);
e. 大腸桿菌 poly(A)聚合酶加尾法:相應的試劑盒有MessageAmp II-bacteria kit (Ambion);與RNA結合蛋白Hfq 等免疫共沉澱法,由於Hfq 能夠高效地結合small RNA,並能輔助它們與靶標mRNA結合,因此常用於small RNA及其靶標mRNA 的研究。
3. 關於文庫構建
針對去除rRNA之後獲得的mRNA進行構建文庫,通常有兩種思路:
a. 先對mRNA結合oligo(dT)進行反轉錄,再針對cDNA進行fragmentation;
b. 先mRNA fragmentation再結合隨機引物進行反轉錄。
這兩種方法獲得的結果會有很多差異:a.藍線;b.紅線。

上圖顯示先針對mRNA進行打斷再進行反轉錄獲得測序reads主要是針對基因本體的;若先反轉錄,尤其是結合oligo(dT)進行反轉錄獲得的測reads對轉錄本3'端具有比較強的偏好性,所以在mRNA-Seq中建議採用先對mRNA打斷再進行反轉錄的文庫構建方法。
根據mRNA文庫構建類別,又分為常規的mRNA文庫構建、均一化文庫構建(引入雙鏈特異性核酸酶)、全長cDNA文庫以及鏈特異性文庫構建(引入dUTP替換合成第二鏈中的dTTP)等,需根據具體的研究目的來選擇,均一化文庫構建可獲得文庫中低丰度表達基因資訊、鏈特異性文庫可獲得正反向鏈上的轉錄本資訊及可變剪下資訊等。
Macrogen 千年基因針對結合NGS平臺測序RNA文庫要求等詳細資訊彙總如下:
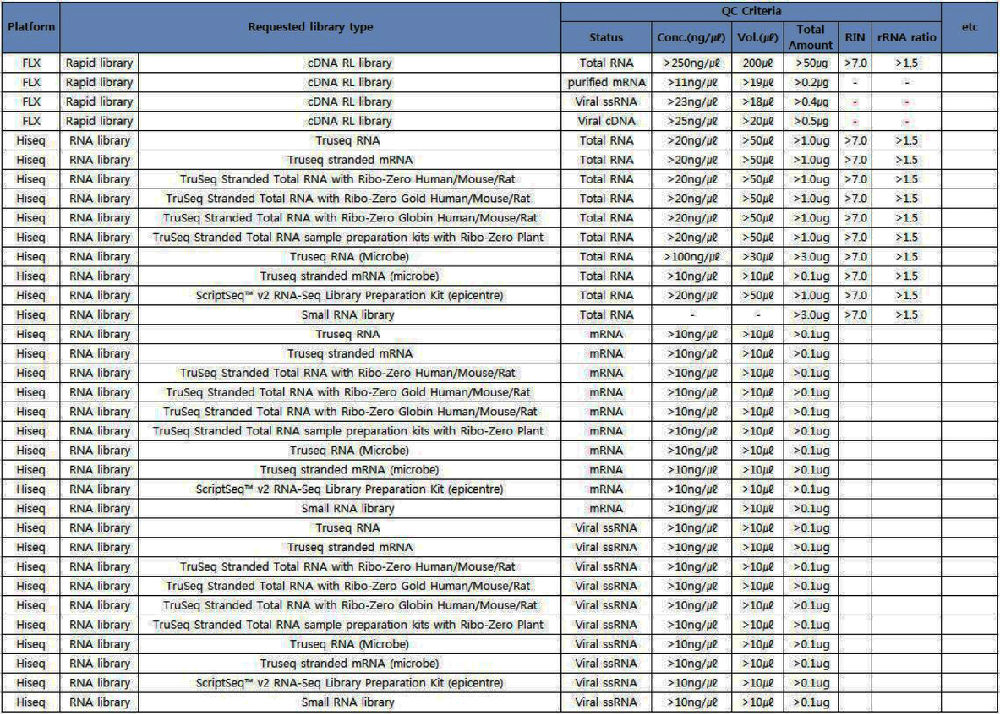
*上述表格針對總RNA以及mRNA、病毒ssRNA的情況均有列出,供參考。
附參考文獻(如有什麼問題歡迎隨時**我[email protected],謝謝!):
1.Influence of RNA extraction methods and library selection schemes on RNA-seq data;
2.IVT-seq reveals extreme bias in RNA-sequencing;
3.Ribosomal RNA depletion for massively parallel bacterial RNA-sequencing applications;
4.Comprehensive comparative analysis of RNA sequencing methods for degraded or low input samples;
7.Prokaryotictranscriptomics: a new view on regulation, physiology and pathogenicity;
8. Efficientand robust RNA-seq process for cultured bacteria and complex communitytranscriptomes;
9. Aperspective: metatranscriptomics as a tool for the discovery of novelbiocatalysts;
11. Globalanalysis of small RNA and mRNA targets of Hfq;
12.Validationof two ribosomal RNA removal methods for microbial metatranscriptomics;
13.RNA-Seq a revolutionary tool for transcriptomics.pdf。