
xgene:疾病相關基因,耳聾,彩色,老年癡呆,帕金森
神經元的傳遞:一個下遊神經元,它接受其上遊神經元的各個突觸傳過來的信號,然而,每個突觸對該下遊神經元的激活權重是不同的。
從神經網絡的本質上說,當人連續、多次遭受失敗的時候,大腦內就會釋放大量的抑制性的神經遞質,會削弱突觸連接。已知有一種叫γ-氨基丁酸 是強的神經抑制劑。
耳聾基因:xgene43
國內常規耳聾基因檢測當中所測的4個耳聾相關基因。一共4個基因。它們分別是:
-
GJB2 :chr13 長臂。隱性遺傳。在耳蝸細胞當中,由GJB2等 6種單體蛋白組成完整的Connexin蛋白,負責細胞內外的鉀離子的電位平衡,並負責傳遞生物電信號。 當GJB2發生突變的時候,就會導致耳蝸的生物電信號的傳遞故障,進而導致耳聾。 突變類型多,黃種人中,最常見的是在第235位缺失一個C堿基(235delC);而在白種人當中,最常見的是在第35位缺少一個G堿基 (35delG)。
-
SLC26A4:別名叫:“PDS基因”,隱性遺傳的。
-
線粒體上的第1555位點A>G突變、和第1494位點C>T突變,突變本身不會導致耳聾,但會對氨基糖苷類的藥物十分敏感,只要使用少量的該類的藥物,就會導致不可逆的耳聾。
-
GJB3:類似GJB2,其538位C>T突變,是一個致病突變。
原理:
一個完整的Connexin蛋白,會在細胞膜上形成一個供離子進出的小孔。
來自2個相鄰細胞的2個完整Connexin蛋白小孔,相互對上,就形成一個通透兩個細胞的離子通道。
當離子通過這個離子通道時,就會在兩個細胞之間引起電流、和電壓的變化。而電流、和電壓的變化達到一定的強度,就會引發神經細胞的興奮,進而達到傳遞信號的目的。
綜合征型的耳聾,和其它的耳聾相關基因
EDN3、EDNRB、MITF、PAX3、SNAI2、和SOX10,這些基因突變,可以會導致Waardenburg綜合征。顯性遺傳病。
EYA1、SIX1、和SIX5,這三個基因當中的一個發生突變,就會引起Branchiootorenal 綜合征,顯性遺傳病。
COL2A1 、或者COL11A1基因突變,會導致Stickler syndrome(綜合征)。顯性遺傳病。
MYO7A、CDH23、USH2A、CLRN1,這些基因突變,會導致Usher syndrome(馬謝爾綜合征)。隱性遺傳疾病。
RHYH、或者PEX7基因突變導致Refsum disease
COL4A3、COL4A4、COL4A5的基因突變,可以導致Alport綜合征。它的特點是:腎臟疾病、聽力下降、眼部異常。
彩色視覺相關基因:xgene44
直接感知彩色光波的,是三種視蛋白。它們分別感知:短、中、長,三種波長的光波。
編碼這三個視蛋白的基因,分別是:
- OPN1SW:短420nm,藍,chr7長臂,
- OPN1MW:中530nm,綠,chr X長臂,與長波只有7個aa差別
- OPN1LW:長560nm,黃綠,chr X長臂,
老年癡呆基因:xgene45
老年癡呆癥,又稱阿爾茨海默癥(Alzheimer‘s disease)是一種慢性的、神經退行性疾病。
主要的表現是:剛開始的時候,記憶力衰退;隨後,語言出現問題、認知出現問題;再發展到生活不能自理,最終死亡。
目前,已知的4個與老年癡呆癥緊密相關的基因,是:
- APP基因:編碼Amyloid beta precursorprotein,即“β澱粉樣前體蛋白”。
- PSEN1基因:與下面基因序列有60%一樣。
- PSEN2基因、
- APOE基因。它編碼一個299個aa的蛋白質。是一種載脂蛋白,它參與乳糜微粒和中間密度脂蛋白代謝。APOE主要有3個等位基因:APOE2、APOE3、和APOE4。差別在於第112位、和第158位的2個氨基酸殘基,是半胱氨酸,還是精氨酸。(可更詳細)
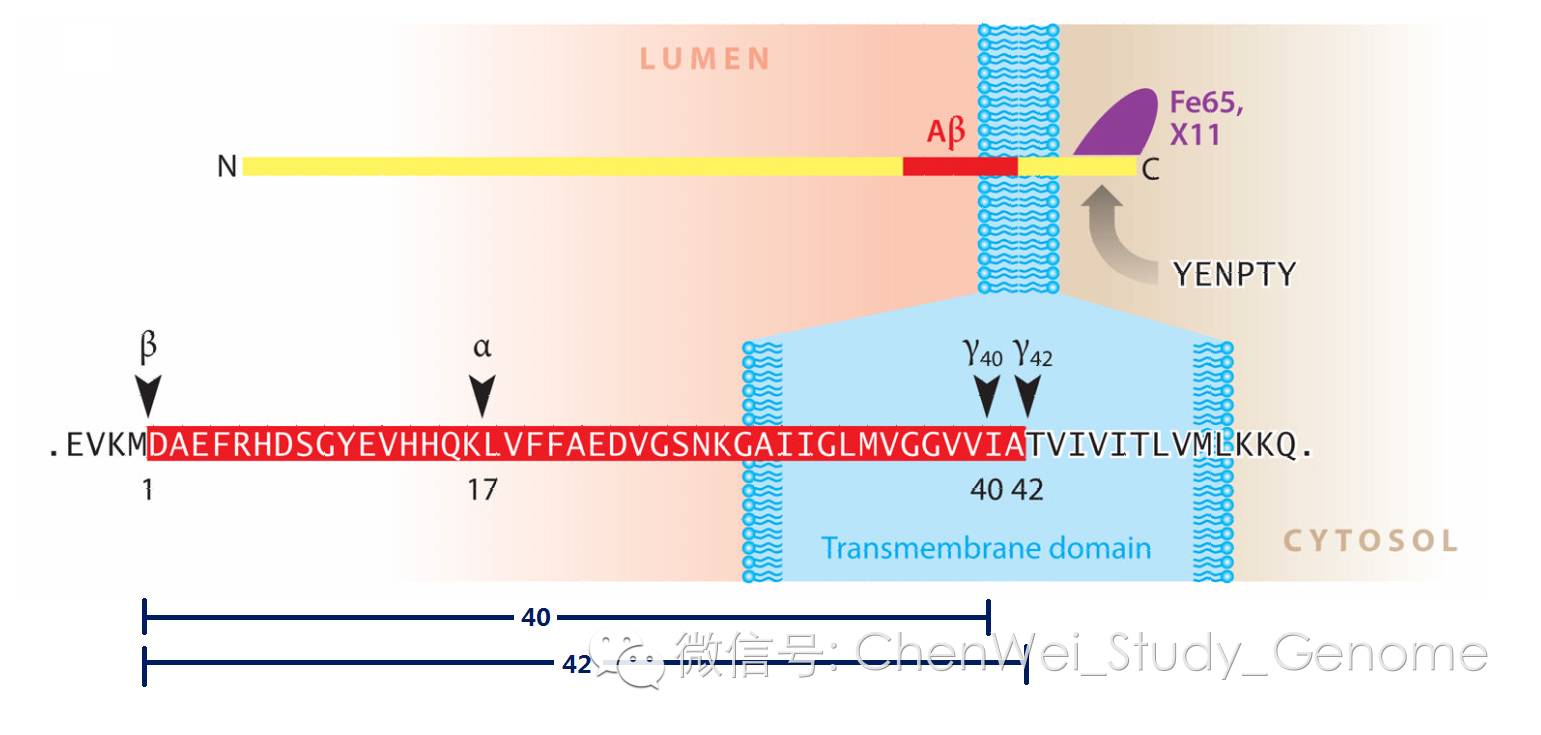
APP基因詳解:

APP蛋白在被翻譯出來之後,就被轉運到細胞膜上,N端的大部分露在細胞膜外,被稱為胞外域,ectodomain。
這部分會被酶給切下來,並參與神經細胞的多種功能。動物實驗表明,這個胞外域肽段,對於動物的認識功能十分重要。
APP蛋白的C端,留在胞質內,它也會被γ分泌酶復合物給切下來,在隨後的細胞內蛋白質降解過程當中,被降解掉。

被切掉了N端、和C端之後的,剩下的中間這一段。這個中間段的N端可以是被α分泌酶在這個位置切斷的,也可以是被β分泌酶在這個位置切斷的。
-
- 如果它的N端是被α分泌酶切斷,然後它的C端是被γ分泌酶切斷,那麽這個中間段就是p3肽。p3肽可以溶解在水中的,不會形成β澱粉樣蛋白沈澱。
- 如果這個中間段的N端是被β分泌酶所切斷,然後C端再被γ分泌酶切一下,那麽,這樣形成的中間段就是β澱粉樣多肽,這會比被α分泌酶所切斷的位置,多出16個aa殘基。 因為這多出的16個氨基酸殘基,讓β澱粉樣多肽在水中的溶解度變得較差。
- 多個β澱粉樣多肽很容易相互聚集起來,形成beta澱粉多肽的寡聚物、和β澱粉樣蛋白沈澱。
psen1 2 詳解
PSEN1和PSEN2,這兩個基因所翻譯出來的蛋白,都是γ分泌酶復合物的組成部分。
分泌酶復合物 在切割APP蛋白的時候 所產生的β澱粉蛋白多肽,可以40個氨基酸殘基的,也可以是42個氨基酸殘基的。
而42個氨基酸殘基的β澱粉蛋白多肽的溶解度更低。更容易發生聚合、沈澱,形成斑塊。所以42個殘基的多肽的毒性更大。
PSEN1、或者PSEN2基因的突變,都會導致γ分泌酶復合物在切割APP蛋白的時候,產生更多的42個氨基酸殘基的多肽。
這就是這兩種基因的突變,會導致早老性癡呆的原因。
β 澱粉樣蛋白 的致病機理
正常情況下,溶解狀態的β澱粉樣多肽、β澱粉樣多肽的寡聚物、和β澱粉樣蛋白沈澱,這三種形式之間,是可以相互轉化的。
在人大腦中,正常情況下β澱粉樣多肽的清除速度是每小時清除8%,當產生多肽的速度大於清除的速度,就會導致β澱粉樣多肽,聚集成寡聚物,或者聚集成β澱粉樣蛋白沈澱。
清除β澱粉樣多肽的酶包括:腦啡肽酶(neprilysin)、胰島素降解酶(insulin degrading enzyme)等。
現在已經發現的,β澱粉樣多肽導致老年癡呆癥的幾種致病機理如下:
-
- Beta澱粉樣多肽聚集成的纖維,這種纖維能夠引發神經元細胞的雕亡
- Beta澱粉樣多肽聚合成的纖維,會堵在神經元的突觸間隙之間,直接影響神經元信號的傳遞
- 溶解的β澱粉多肽,會影響tau蛋白的(被)切割、和磷酸化
而tau蛋白的(被)切割和磷酸化,是神經元死亡 並形成神經元纖維纏結的重要原因。
關於老年癡呆癥,目前的科學研究,還只了解了一部分的發病機理。
帕金森疾病
主要臨床表現為:靜止性震顫、運動遲緩、肌張力高、走路姿勢、和站立姿勢都出現異常。
同時患者還可能伴有:抑郁、便秘、睡眼障礙、和癡呆等癥狀。病情會逐步加重。病人從發病到死亡,可能會經歷10~20年的時間。
在所有的神經退行性疾病當中,老年癡呆癥的發病率排第1,帕金森病的發病率排第2。
已經發現的與帕金森病相關的基因有30多個。重點談其中的5個:
- SNCA、
- LRRK2、
- MAPT、
- PARK2、
- PINK1:
1,SNCA基因突變:一般是認為SNCA基因的點突變,會導致α-突觸核蛋白更加容易聚集成Lewy小體;或SNCA基因拷貝數異常增加,會直接增加α-突觸核蛋白的數量,過多的α-突觸核蛋白,就聚集成Lewy小體。。。而Lewy小體阻礙了多巴胺的代謝,和神經元的正常功能,導致神經元的死亡。
2,現在一般認為,LRRK2基因所表達的蛋白的功能,主是是參與細胞骨架的組裝,以及參與蛋白與蛋白之間的相互作用。顯性遺傳。。
3,MAPT基因,就是“微管相關蛋白tau”,編碼TAU蛋白。TAU蛋白,是組成微管的一個重要組成部分。已經發現了40多種MAPT基因的突變,與帕金森病的發病相關。
4,PARK2基因,又名Parkin基因。它編碼一個泛素連接酶。
泛素連接酶,會把細胞內的蛋白質連接上泛素,讓蛋白質進入泛素降解途徑,把蛋白質給降解掉。
當PARK2(Parkin)基因發生突變,就不能把該降解的蛋白質連上泛素,這樣,多余的蛋白質就不能被降解,而會堆積在細胞內。
5,PINK1基因有“維持線粒體功能”的作用。隱性遺傳的。
在正常情況下,PINK1的蛋白可以被蛋白酶切斷,並進入線粒體的內膜,發揮正常作用,而線粒體的內膜也可以保持正常的極化。
而當PINK1的蛋白有突變的時候,它不能被蛋白酶切斷,也就不能進入線粒體的內膜,這樣線粒體的內膜就會去極化,最終導致神經元細胞的死亡。
現在已經在PINK1上,發現了70多種突變,會導致帕金森病。
一般認為,它們是通過參與以下這4個生物通路,導致帕金森病的:
-
1,突觸的神經信號傳遞
-
2,細胞內的物質運輸
-
3,溶酶體的自噬作用
-
4,線粒體的代謝
xgene:疾病相關基因,耳聾,彩色,老年癡呆,帕金森