
hisat2+stringtie+ballgown
阿新 • • 發佈:2018-11-09
hisat2+stringtie+ballgown
Posted on 2016年11月25日早在去年九月,我就寫個博文說 RNA-seq流程需要進化啦! http://www.bio-info-trainee.com/1022.html ,主要就是進化成hisat2+stringtie+ballgown的流程,但是我一直沒有系統性的講這個流程,因為我覺真心木有用。我只用了裡面的hisat來做比對而已!但是群裡的小夥伴問得特別多,我還是勉為其難的寫一個教程吧,你們之間拷貝我的程式碼就可以安裝這些軟體的!然後自己找一個測試資料,我的指令碼很容易用的!
## Download and install HISAT # https://ccb.jhu.edu/software/hisat2/index.shtml cd ~/biosoft mkdir HISAT && cd HISAT #### readme: https://ccb.jhu.edu/software/hisat2/manual.shtml wget ftp://ftp.ccb.jhu.edu/pub/infphilo/hisat2/downloads/hisat2-2.0.4-Linux_x86_64.zip unzip hisat2-2.0.4-Linux_x86_64.zip ln -s hisat2-2.0.4 current ## ~/biosoft/HISAT/current/hisat2-build ## ~/biosoft/HISAT/current/hisat2 ## Download and install StringTie ## https://ccb.jhu.edu/software/stringtie/ ## https://ccb.jhu.edu/software/stringtie/index.shtml?t=manual cd ~/biosoft mkdir StringTie && cd StringTie wget http://ccb.jhu.edu/software/stringtie/dl/stringtie-1.2.3.Linux_x86_64.tar.gz tar zxvf stringtie-1.2.3.Linux_x86_64.tar.gz ln -s stringtie-1.2.3.Linux_x86_64 current # ~/biosoft/StringTie/current/stringtie
while read id do sample=$(echo $id |cut -d" " -f 1 ) file1=$(echo $id |cut -d" " -f 2 ) file2=$(echo $id |cut -d" " -f 3 ) echo $sample echo $file1 echo $file2 ~/biosoft/HISAT/current/hisat2 -p 4 --dta -x ~/reference/index/hisat/hg19/genome -1 $file1 -2 $file2 -S $sample.hisat2.hg19.sam 2>$sample.hisat2.hg19.log & done <$1
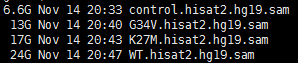
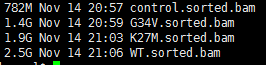
while read id do file=$(basename $id ) sample=${file%%.*} echo $id $sample nohup samtools sort [email protected] 4 -o ${sample}.sorted.bam $id & done <$1最新版的samtools已經可以直接把sam檔案變成排序好的bam檔案啦~~~~
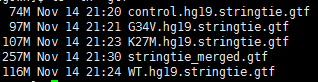
while read id do file=$(basename $id ) sample=${file%%.*} echo $id $sample nohup ~/biosoft/StringTie/current/stringtie -p 4 -G ~/reference/gtf/gencode/gencode.v25lift37.annotation.gtf -o $sample.hg19.stringtie.gtf -l $sample $id & done <$1stringTie的用法就是這樣咯。沒什麼好講的
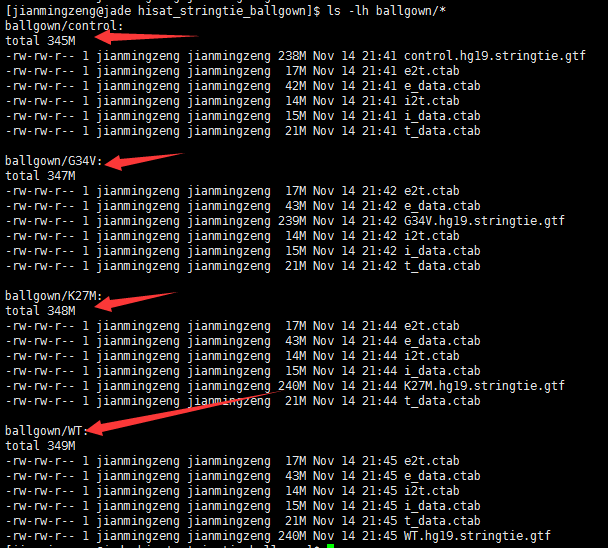
This entry was posted in 轉錄組軟體 and tagged ballgown, hisat2, StringTie, 轉錄組 by ulwvfje. Bookmark the permalink.