
使用clusterProfiler進行KEGG富集分析
歡迎關注微信公眾號《生信修煉手冊》!
KEGG pathway是最常用的功能註釋資料庫之一,可以利用KEGG 的API獲取一個物種所有基因對應的pathway註釋,human對應的API 連結如下
通過該連結可以獲得以下內容
path:hsa00010 hsa:10327
path:hsa00010 hsa:124
path:hsa00010 hsa:125
第一列為pathway編號,第二列為基因編號。這裡只提供了pathway編號,我們還需要pathway對應的描述資訊,同樣也可以通過以下API連結得到
通過該連結可以獲得如下內容
path:map00010 Glycolysis / Gluconeogenesis path:map00020 Citrate cycle (TCA cycle) path:map00030 Pentose phosphate pathway path:map00040 Pentose and glucuronate interconversions path:map00051 Fructose and mannose metabolism
第一列為pathway編號,第二列為具體的描述資訊。需要注意的是,pathway是一個跨物種的概念,原始的pathway編號為map或者ko加數字,對於特定物種,改成物種對應的三字母縮寫, 比如human對應hsa, 所有擁有pathway資訊的物種和對應的三字母縮寫見如下連結
clusterProfiler也是通過KEGG API去獲取物種對應的pathway註釋,對於已有pathway註釋的物種,我們只需要知道對應的三字母縮寫, clusterProfiler就會聯網自動獲取該物種的pathway註釋資訊。
和GO富集分析類似,對於KEGG的富集分析也包含以下兩種
1. Over-Representation Analysis
過表達分析其實就是費舍爾精確檢驗,分析的程式碼如下
ego <- enrichKEGG(
gene = gene,
keyType = "kegg",
organism = 'hsa',
pvalueCutoff = 0.05,
pAdjustMethod = "BH",
qvalueCutoff = 0.05
)
我們只需要提供差異基因的列表和物種對應的三字母縮寫,預設基因ID為kegg gene id, 通過keyType引數指定,也可以是ncbi-geneid, ncbi-proteind, uniprot。
不同型別ID的轉換也是通過KEGG API實現的,比如hsa
hsa:1 ncbi-geneid:1
hsa:100009667 ncbi-geneid:100009667
hsa:100009676 ncbi-geneid:100009676
hsa:10 ncbi-geneid:10
hsa:100 ncbi-geneid:100
在clusterProfiler中,可以通過bitr_kegg函式,呼叫KEGG API, 來實現ID 轉換功能,示例如下
bitr_kegg(
"1",
fromType = "kegg",
toType = 'ncbi-proteinid',
organism='hsa')
2. Gene Set Enrichment Analysis
對應的函式為gseKEGG, 用法如下
kk <- gseKEGG(
geneList = gene,
keyType = 'kegg',
organism = 'hsa',
nPerm = 1000,
minGSSize = 10,
maxGSSize = 500,
pvalueCutoff = 0.05,
pAdjustMethod = "BH"
)
對於pathway資料庫中沒有的物種,也支援讀取基因的pathway註釋檔案,然後進行分析,註釋檔案的格式如下
| GeneId | KEGG | Description |
|---|---|---|
| 1 | ko:00001 | spindle |
| 2 | ko:00002 | mitotic spindle |
| 3 | ko:00003 | kinetochore |
只需要3列資訊即可,第一列為geneID, 第二列為基因對應的pathway編號,第三列為pathway的描述資訊。這3列的順序是無所謂的, 只要包含這3種資訊就可以了。
讀取該檔案,進行分析的程式碼如下
data <- read.table(
"pathway_annotation.txt",
header = T,
sep = "\t")
go2gene <- data[, c(2, 1)]
go2name <- data[, c(2, 3)]
# 費舍爾精確檢驗
x <- enricher(
gene,
TERM2GENE = go2gene,
TERM2NAME = go2name)
# GSEA富集分析
x <- GSEA(
gene,
TERM2GENE = go2gene,
TERM2NAME = go2name)
對於KEGG富集分析的結果,clusterProfiler提供了以下幾種視覺化策略
1. barplot
用散點圖展示富集到的pathways,用法如下
barplot(kk, showCategory = 10)
生成的圖片如下
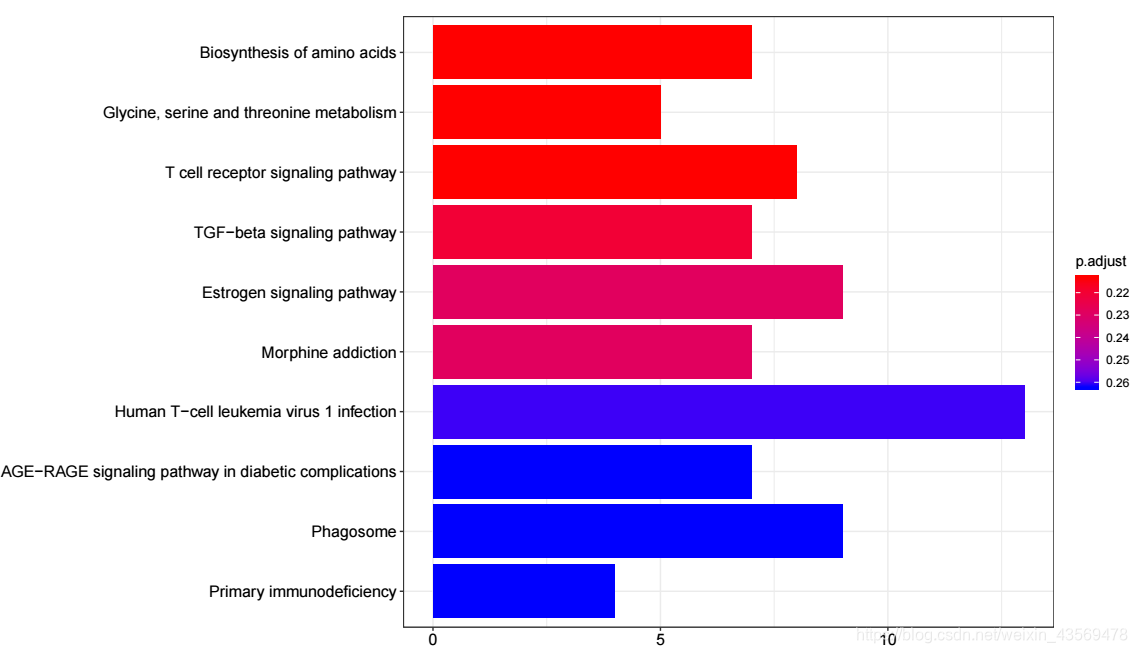 橫軸為該pathway的差異基因個數,縱軸為富集到的pathway的描述資訊,
橫軸為該pathway的差異基因個數,縱軸為富集到的pathway的描述資訊, showCategory指定展示的pathway的個數,預設展示顯著富集的top10個,即p.adjust最小的10個。注意的顏色對應p.adjust值,從小到大,對應藍色到紅色。
2. dotplot
用散點圖展示富集到的pathways,用法如下
dotplot(kk, showCategory = 10)
生成的圖片如下
 橫軸為
橫軸為GeneRatio, 代表該pathway下的差異基因個數佔差異基因總數的比例,縱軸為富集到的pathway的描述資訊, showCategory指定展示的pathway的個數,預設展示顯著富集的top10個,即p.adjust最小的10個。圖中點的顏色對應p.adjust的值,從小到大,對應藍色到紅色,大小對應該GO terms下的差異基因個數,個數越多,點越大。
3. emapplot
對於富集到的pathways之間的基因重疊關係進行展示,如果兩個pathway的差異基因存在重疊,說明這兩個節點存在overlap關係,在圖中用線條連線起來,用法如下
emapplot(kk, showCategory = 30)
生成的圖片如下
 每個節點是一個富集到的pathway, 預設畫top30個富集到的pathways, 節點大小對應該pathway下富集到的差異基因個數,節點的顏色對應p.adjust的值,從小到大,對應藍色到紅色。
每個節點是一個富集到的pathway, 預設畫top30個富集到的pathways, 節點大小對應該pathway下富集到的差異基因個數,節點的顏色對應p.adjust的值,從小到大,對應藍色到紅色。
4. cnetplot
對於基因和富集的pathways之間的對應關係進行展示,如果一個基因位於一個pathway下,則將該基因與pathway連線,用法如下
cnetplot(kk, showCategory = 5)
生成的圖片如下
 圖中灰色的點代表基因,黃色的點代表富集到的pathways, 預設畫top5富集到的pathwayss, pathways節點的大小對應富集到的基因個數。
圖中灰色的點代表基因,黃色的點代表富集到的pathways, 預設畫top5富集到的pathwayss, pathways節點的大小對應富集到的基因個數。
5. browseKEGG
在pathway通路圖上標記富集到的基因,程式碼如下
browseKEGG(kk, "hsa04934")
會給出一個url連結,示例如下
在瀏覽器中開啟會看到如下所示的圖片
 富集到的差異基因會用紅色方框表示,更多用法和細節請參考官方文件。
富集到的差異基因會用紅色方框表示,更多用法和細節請參考官方文件。
掃描關注微訊號,更多精彩內容等著你!
