
單細胞分析實錄(5): Seurat標準流程
阿新 • • 發佈:2021-01-06
前面我們已經學習了單細胞轉錄組分析的:[使用Cell Ranger得到表達矩陣](https://www.jianshu.com/p/5ace1c5b18b7)和[doublet檢測](https://www.jianshu.com/p/64976eb6725d),今天我們開始Seurat標準流程的學習。這一部分的內容,網上有很多帖子,基本上都是把[Seurat官網PBMC的例子](https://satijalab.org/seurat/v3.2/pbmc3k_tutorial.html)重複一遍,這回我換一個數據集,細胞型別更多,同時也會加入一些實際分析中很有用的技巧。
### 1. 匯入資料,建立Seurat物件
```
library(Seurat)
library(tidyverse)
testdf=read.table("test_20210105.txt",header = T,row.names = 1)
test.seu=CreateSeuratObject(counts = testdf)
```
看一下長什麼樣子
```
> test.seu
An object of class Seurat
33538 features across 6746 samples within 1 assay
Active assay: RNA (33538 features)
```
測試資料有33538個基因,6746個細胞。除此之外,還要關注一下另外兩層資訊:[email protected]這個資料框用來儲存元資料,每一個細胞都有多個屬性;test.seu[["RNA"]]@counts這個稀疏矩陣用來儲存原始UMI表達矩陣。
```
> head([email protected])
orig.ident nCount_RNA nFeature_RNA
A_AAACCCAAGGGTCACA A 3714 1151
A_AAACCCAAGTATAACG A 1855 816
A_AAACCCAGTCTCTCAC A 1530 823
A_AAACCCAGTGAGTCAG A 11145 1087
A_AAACCCAGTGGCACTC A 2289 834
A_AAACGAAAGCCAGAGT A 3714 990
> test.seu[["RNA"]]@counts[1:4,1:4]
4 x 4 sparse Matrix of class "dgCMatrix"
A_AAACCCAAGGGTCACA A_AAACCCAAGTATAACG A_AAACCCAGTCTCTCAC A_AAACCCAGTGAGTCAG
MIR1302-2HG . . . .
FAM138A . . . .
OR4F5 . . . .
AL627309.1 . . . .
```
### 2. 簡單過濾
接下來,我們根據每個細胞內部`線粒體基因表達佔比`、`檢測到的基因數`、`檢測的UMI總數`這三個方面來對細胞進行簡單的過濾。
先計算細胞內線粒體基因表達佔比,類似的核糖體基因(大多為RP開頭)也能這樣計算,還要注意不要將線粒體基因的`MT-`寫成了`MT`,不然就把別的基因也算進去了:
```
test.seu[["percent.mt"]] <- PercentageFeatureSet(test.seu, pattern = "^MT-") #正則表示式,表示以MT-開頭;test.seu[["percent.mt"]]這種寫法會在meta.data矩陣加上一列
```
這裡我已經根據預先設定好的閾值過濾了,程式碼如下
```
test.seu <- subset(test.seu, subset = nCount_RNA > 1000 &
nFeature_RNA < 5000 &
percent.mt < 30 &
nFeature_RNA > 600)
```
過濾之後的數值分佈如下,用到`VlnPlot()`函式,該繪圖函式裡面的feature引數可以是meta.data矩陣的某一列,也可以是某一個基因,很多文章都用這種圖展示marker gene
```
VlnPlot(test.seu,features = c("nCount_RNA", "nFeature_RNA", "percent.mt"))
```

>nFeature_RNA/nCount_RNA不能太小(空液滴),不能太大(doublet、測序技術限制), 而且閾值設定要綜合多個樣本來看,像下面這樣
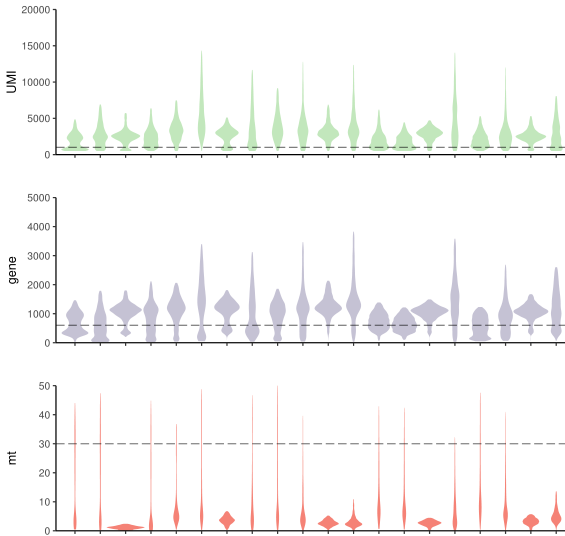
> 一般在CD45陰性的細胞中percent.mt的閾值大一些,50%也看過幾次了
### 3. 標準化,消除文庫大小的影響
> 如何標準化:LogNormalize: Feature counts for each cell are divided by the total counts for that cell and multiplied by the scale.factor. This is then natural-log transformed using log1p.(先相除,再求對數)
```
test.seu <- NormalizeData(test.seu, normalization.method = "LogNormalize", scale.factor = 10000)
```
標準化之後的矩陣儲存在test.seu[["RNA"]]@data
```
> test.seu[["RNA"]]@data[1:4,1:4]
4 x 4 sparse Matrix of class "dgCMatrix"
A_AAACCCAAGGGTCACA A_AAACCCAAGTATAACG A_AAACCCAGTCTCTCAC A_AAACCCAGTGAGTCAG
MIR1302-2HG . . . .
FAM138A . . . .
OR4F5 . . . .
AL627309.1 . . . .
```
### 4. 找Variable基因
因為單細胞表達矩陣很稀疏(很多0),選high variable基因的目的可以找到包含資訊最多的基因(很多基因的表達差不多都是0),同時極大提升軟體執行速度
```
test.seu <- FindVariableFeatures(test.seu, selection.method = "vst", nfeatures = 2000)
```
這些基因儲存在VariableFeatures(test.seu),有時候可能需要人為指定high variable基因,可以這樣:
```
VariableFeatures(test.seu)="specific genes"
```
### 5. 歸一化表達矩陣
(基於前面得到的data矩陣)
這一步之後,所有基因的表達值的分佈就差不多了,不然表達值不在一個數量級,對後續降維聚類影響挺大。新的矩陣儲存在test.seu[["RNA"]]@scale.data裡面。
```
test.seu <- ScaleData(test.seu, features = rownames(test.seu))
```
預設只對上一步選出來的基因scale,這裡調整為所有基因,是為了方便以後畫熱圖,畫熱圖一般會用scale之後的z-score
### 6. 降維聚類
(基於前面得到的high variable基因的scale矩陣)
```
test.seu <- RunPCA(test.seu, npcs = 50, verbose = FALSE)
test.seu <- FindNeighbors(test.seu, dims = 1:30)
test.seu <- FindClusters(test.seu, resolution = 0.5)
test.seu <- RunUMAP(test.seu, dims = 1:30)
test.seu <- RunTSNE(test.seu, dims = 1:30)
```
Run開頭的函式降維,Find開頭的函式聚類,一般就這幾步,相對固定。PCA將原來2000維的資料降到50維,dims引數表示使用多少個主成分(一般20左右就可以了,多幾個少幾個對結果影響不大),resolution引數表達聚類的解析度,這個值大於0,一般都是在0-1範圍裡面調整,越大得到的cluster越多,這個值可以反覆調整,並不會改變降維的結果(也就是tsne、umap圖的二維座標)。
這一步之後的資料是這樣的
```
> test.seu
An object of class Seurat
33538 features across 6746 samples within 1 assay
Active assay: RNA (33538 features)
3 dimensional reductions calculated: pca, umap, tsne
# 幾種降維方式都會呈現出來
```
聚類之後多了兩列,RNA_snn_res.0.5記錄了你用的解析度,最終的聚類結果儲存在seurat_clusters中
```
> head([email protected])
orig.ident nCount_RNA nFeature_RNA percent.mt RNA_snn_res.0.5
A_AAACCCAAGGGTCACA A 3714 1151 9.585353 8
A_AAACCCAAGTATAACG A 1855 816 12.776280 0
A_AAACCCAGTCTCTCAC A 1530 823 14.248366 12
A_AAACCCAGTGAGTCAG A 11145 1087 2.853297 4
A_AAACCCAGTGGCACTC A 2289 834 15.640017 3
A_AAACGAAAGCCAGAGT A 3714 990 5.654281 0
seurat_clusters
A_AAACCCAAGGGTCACA 8
A_AAACCCAAGTATAACG 0
A_AAACCCAGTCTCTCAC 12
A_AAACCCAGTGAGTCAG 4
A_AAACCCAGTGGCACTC 3
A_AAACGAAAGCCAGAGT 0
```
### 7. tsne/umap展示結果
```
library(cowplot)
test.seu$patient=str_replace(test.seu$orig.ident,"_.*$","")
p1 <- DimPlot(test.seu, reduction = "tsne", group.by = "patient", pt.size=0.5)
p2 <- DimPlot(test.seu, reduction = "tsne", group.by = "ident", pt.size=0.5, label = TRUE,repel = TRUE) #後面兩個引數用來新增文字標籤
p3 <- DimPlot(test.seu, reduction = "umap", group.by = "patient", pt.size=0.5)
p4 <- DimPlot(test.seu, reduction = "umap", group.by = "ident", pt.size=0.5, label = TRUE,repel = TRUE)
fig_tsne <- plot_grid(p1, p2, labels = c('patient','ident'),align = "v",ncol = 2)
ggsave(filename = "tsne.pdf", plot = fig_tsne, device = 'pdf', width = 30, height = 15, units = 'cm')
fig_umap <- plot_grid(p3, p4, labels = c('patient','ident'),align = "v",ncol = 2)
ggsave(filename = "umap.pdf", plot = fig_umap, device = 'pdf', width = 30, height = 15, units = 'cm')
```
ident表示每個細胞的標籤,聚類之後就是聚類的結果,在一些特定場景可以更換。
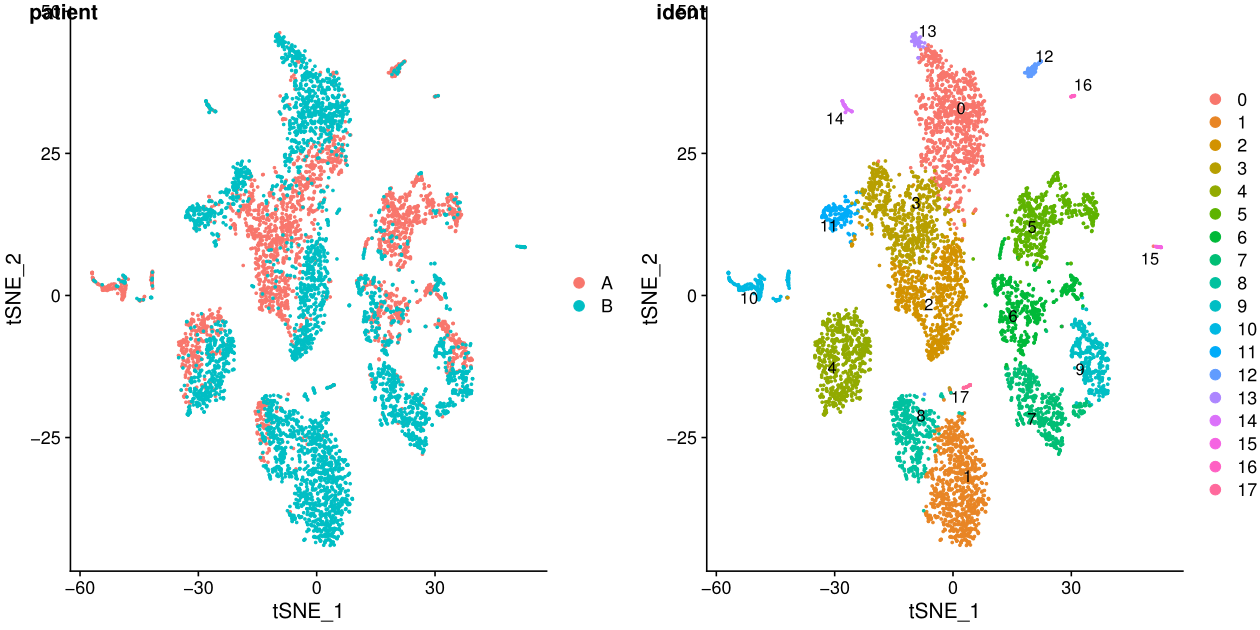
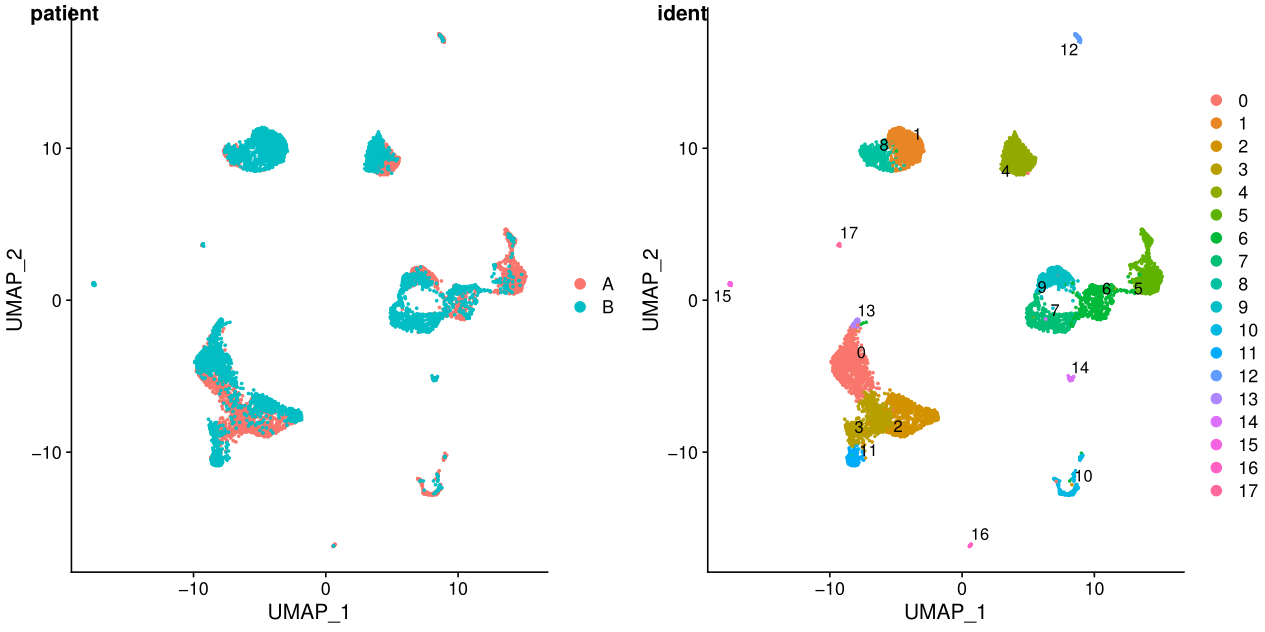
在umap圖中,cluster之間的距離更明顯
***
從上面的圖可以看出不同樣本其實是有批次效應的,下一講我會介紹兩種去批次效應的方法。
> 因水平有限,有錯誤的地方,歡迎批評