
單細胞分析實錄(7): 差異表達分析/細胞型別註釋
阿新 • • 發佈:2021-01-07
> 前面已經講解了:
> [單細胞分析實錄(1): 認識Cell Hashing](https://www.jianshu.com/p/02b70bda5d0a)
> [單細胞分析實錄(2): 使用Cell Ranger得到表達矩陣](https://www.jianshu.com/p/5ace1c5b18b7)
> [單細胞分析實錄(3): Cell Hashing資料拆分](https://www.jianshu.com/p/d5aaecc76214)
> [單細胞分析實錄(4): doublet檢測](https://www.jianshu.com/p/64976eb6725d)
> [單細胞分析實錄(5): Seurat標準流程](https://www.jianshu.com/p/ce6205fe1ff9)
> [單細胞分析實錄(6): 去除批次效應/整合資料](https://www.jianshu.com/p/a7348a5f1ae7)
這一節我們進入細胞型別註釋的學習,總的來說,兩條路線,手動註釋和軟體註釋。我在實際處理的過程中,主要還是手動註釋,軟體的註釋結果只作為一個參考,來輔助證實手動註釋的結果是準確無誤的。
相信看過幾篇單細胞文獻的小夥伴基本都會知道幾種常見細胞大類的marker,我們在註釋的時候用這些marker就可以基本確定細胞大類了。另外,以SingleR為例,對於細胞大類的註釋還算準確,當然也沒有到很準確的程度,我試過更換SingleR裡面不同的參考集,最後得到的大類註釋結果一致性不到80%。對於細胞小類的註釋,軟體就更加無法勝任了,我幾乎沒有看過高分的文獻會用SingleR的小類註釋結果。當然不排除隨著單細胞研究的普及和深入,以後能有更準確的軟體出現。
接下來,我以上一節的資料為例,走一遍我的分析流程。
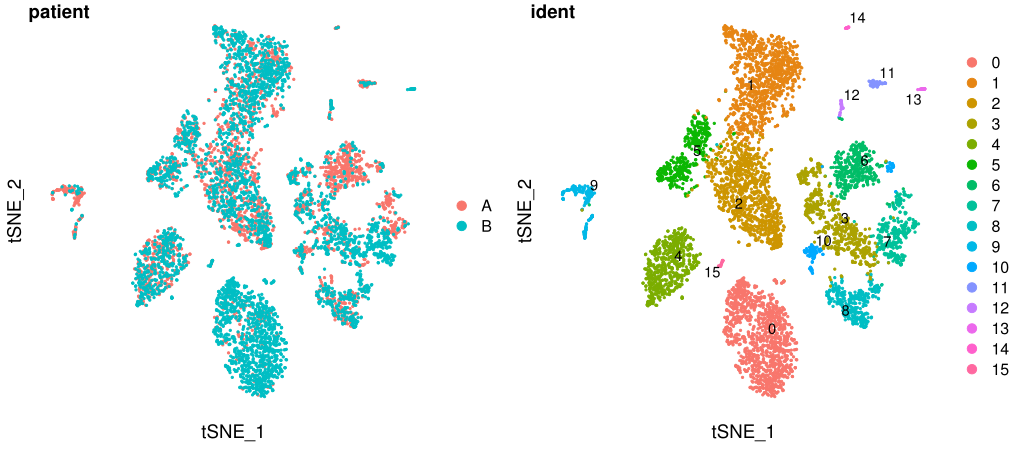
之前的資料呈現出了16個cluster,至於resolution引數的選擇,我的原則是能在降維圖上分開的細胞亞群,能有它自己的cluster label,並且成團較好,較緊緻的一群細胞沒有必要再強行分群(比如上圖的第4群)。
這16個cluster不一定都會用到,因為doublet、細胞數太少等原因,可能還得去掉。
我推薦用常見的marker先區分一下,大概能知道cluster對應什麼型別。這裡用到的marker都是在文獻裡面經常出現的,我自己也整理了一份更全一點的細胞型別marker清單,有需要的可以在公眾號後臺說明。
```
celltype_marker=c(
"EPCAM",#上皮細胞 epithelial
"PECAM1",#內皮細胞 endothelial
"COL3A1",#成纖維細胞 fibroblasts
"CD163","AIF1",#髓系細胞 myeloid
"CD79A",#B細胞
"JCHAIN",#漿細胞 plasma cell
"CD3D","CD8A","CD4",#T細胞
"GNLY","NKG7",#NK細胞
"PTPRC"#免疫細胞
)
VlnPlot(test.seu,features = celltype_marker,pt.size = 0,ncol = 2)
ggsave(filename = "marker.png",device = "png",width = 44,height = 33,units = "cm")
```
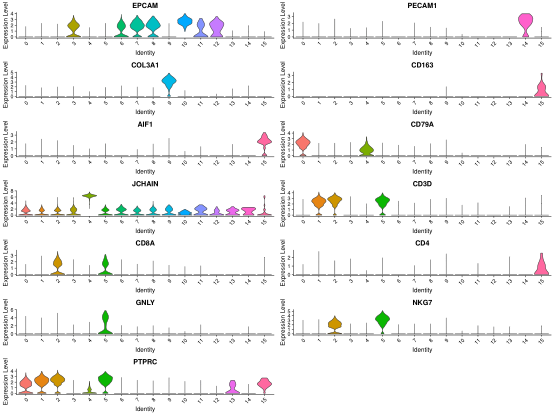
結合對應的關係,初步確認細胞型別如下,需要說明的是,NK細胞和T細胞不是很能區分開,一些文獻也是直接把這兩個當做一個大群來做的,此處我根據GNLY基因確定第5個cluster為NK細胞,是否正確我們後面再看。
```
#免疫細胞
0: B_cell
4: Plasma_cell
1 2: T_cell
5: NK_cell
13: Unknown
#非免疫細胞
3 6 7 8 10 11 12: Epithelial
14: Endothelial
9: Fibroblasts
#doublet
15: Doublet (Myeloid+CD4)
```
以上根據經典的marker初步確定了細胞大類,接著我們找差異基因,看看找出來的每一個cluster的差異基因是不是和前面鑑定的型別一致。
### 1. 找差異基因
```
markers <- FindAllMarkers(test.seu, logfc.threshold = 0.25, min.pct = 0.1,
only.pos = TRUE, test.use = "wilcox")
markers_df = markers %>% group_by(cluster) %>% top_n(n = 500, wt = avg_logFC)
write.table(markers_df,file="test.seu_0.5_logfc0.25_markers500.txt",quote=F,sep="\t",row.names=F,col.names=T)
```
FindAllMarkers()這個函式會將cluster中的某一類和剩下的那些類比較,來找差異基因。與其對應的有個FindMarkers()函式,可以指定哪兩個cluster對比。logfc.threshold表示logfc的閾值,這裡有兩個地方需要注意:一是Seurat裡面的logfc計算公式很特別,並不是我們平常在bulk裡面那樣算均值,相除,求log,但其實也不要糾結怎麼算的,只需知道這是表示倍數的一個指標即可;二是如果想畫火山圖,這個閾值可以設為0,不然最後畫出來的火山圖中間會缺一段,我們完全可以把全部基因先拿出來,畫火山圖,再根據p value, logFC這些閾值自己過濾。
min.pct表示基因在多少細胞中表達的閾值,only.pos = TRUE表示只求高表達的基因,test.use表示檢測差異基因所用的方法。
第2行程式碼,根據avg_logFC這一列把前500個差異基因取出來,小於500個時,有多少保留多少。
事實上,不必在意我們保留多少個差異基因,實際用到的,也就前面十幾(幾十)個基因。
最終得到的差異基因list如下:
```
> head(markers_df)
# A tibble: 6 x 7
# Groups: cluster [1]
p_val avg_logFC pct.1 pct.2 p_val_adj cluster gen