
基因晶片(Affymetrix)分析5:聚類分析
聚類分析的目的是為了獲得合適的分類。但怎麼才算“合適”?這也是個大問題。典型的做法是先設定一個閾值,再使用這個閾值擷取系統樹,能截出幾個系統樹分枝就是幾個簇。通過hclust型別資料擷取系統樹分枝分3步進行:
- 用cutree函式擷取hclust資料
- 獲取各分枝的基因名稱並通過基因名稱取距離矩陣的子集
- 使用距離矩陣子集重構系統樹
後兩步都很熟悉了,主要看看第1步。cutree函式引數h是我們設定的擷取閾值,得到的資料是帶名稱的向量:元素名稱是基因名,元素的值是系統樹分枝編號。
clust.fc <- hclust(dist.fc, method="complete") branches.fc<- cutree(clust.fc, h=0.4) #看看資料頭的樣子 head(branches.fc)
## 254818_at 245998_at 265119_at 256114_at 254805_at 251775_s_at ## 1 2 2 2 1 2
#截取出的分枝和各分枝基因數 table(branches.fc)
## branches.fc ## 1 2 3 4 5 6 7 8 9 10 ## 1960 2415 3035 845 592 1410 1655 1459 1574 1060
接下來可以取距離矩陣子集並重構第系統樹分枝。 你還可以在原系統樹上通過畫方框的方法標出各分枝:
br1 <- names(branches.fc[branches.fc==1]) dist.br1 <- as.dist(as.matrix(dist.fc)[br1,br1]) clust.br1 <- hclust(dist.br1, method="complete") br3 <- names(branches.fc[branches.fc==3]) dist.br3 <- as.dist(as.matrix(dist.fc)[br3,br3])clust.br3 <- hclust(dist.br3, method="complete") par(mfrow=c(1,3)) par(mar=c(1,4,2,0)) plot(clust.br1, hang=-1, labels=FALSE, main='branch 1', sub="") plot(clust.br3, hang=-1, labels=FALSE, main='branch 3', sub="") plot(clust.fc, hang=-1, labels=FALSE, main='clust.fc', sub="") rect.hclust(clust.fc, h=0.4, border="red")

但從上圖框出的分枝大小和前面的分枝基因數來看,各分枝並不是按圖上橫座標的順序進行排列的。
如果你的目的是獲得基因列表,那麼上面的方法就可以了;如果想獲得更加美觀點的圖形,可以將hclust產生的結果轉為系統樹(dendrogram)型別。系統樹型別的資料也可以直接使用plot函式繪圖,引數比hclust繪圖更多:
dend.fc <- as.dendrogram(clust.fc, hang=-1) par(mfrow=c(1,1)) par(mar=c(2,1,1,1)) plot(dend.fc, leaflab="none", horiz=T, edgePar = list(col="seagreen", lwd=2), main="Plot dendrogram data")
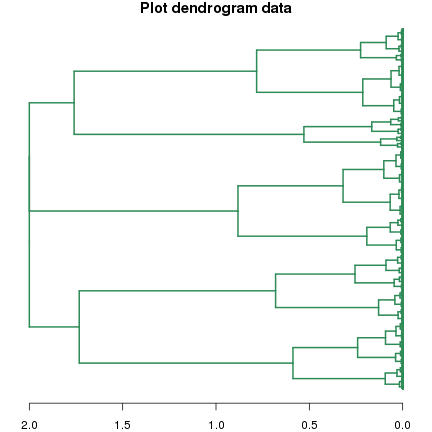
而且還可以使用cut函式擷取系統樹。cut( )函式通過設定height引數(閾值)來擷取系統樹,獲得的結果是列表結構,有名稱為upper和lower的兩個列表成員:
cut.fc <- cut(dend.fc, h=0.4) class(cut.fc)
## [1] "list"
再看upper和lower的型別,發現upper是系統樹型別,而lower是列表。把upper系統樹plot處理你就知道它是什麼了:
class(cut.fc$upper); class(cut.fc$lower);
## [1] "list"
plot(cut.fc$upper, leaflab="none", horiz=T, edgePar = list(col="seagreen", lwd=2), main="cut.fc$upper")
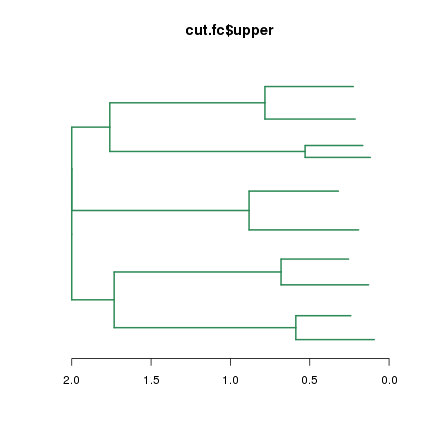
再看看lower列表,它有10個列表元素,每個都是系統樹資料,我們當然也可以plot這些系統樹分枝:
cnames <- NULL for(i in 1:length(cut.fc$lower)) cnames <- c(cnames, (class(cut.fc$lower[[i]]))) cnames
## [1] "dendrogram" "dendrogram" "dendrogram" "dendrogram" "dendrogram" ## [6] "dendrogram" "dendrogram" "dendrogram" "dendrogram" "dendrogram"
plot(cut.fc$lower[[1]],leaflab="none", horiz=T, main="cut.fc$lower[[1]]")
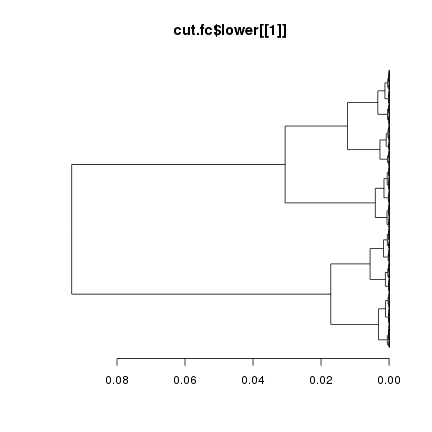
事實上我們可以通過另外的方法看到這10個分枝:
plot(dend.fc, leaflab="none", horiz=T, edgePar = list(col="seagreen", lwd=2), xlim=c(0.4,0))
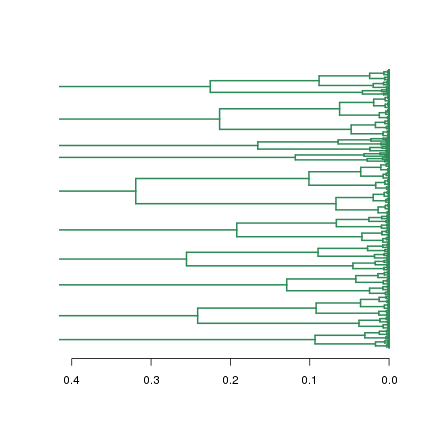
請看cut.fc$lower1是不是就是最下面的分支?從這也可以看出cut.fc$lower中各項的排列順序,接下來再分析各個分支就有譜了。dendrogram型別資料要獲得基因名稱列表還需返回到hclust資料型別,然後通過hclust的labels屬性獲得基因列表。因為cutree和cut函式產生的分枝編號不能對應起來,這一步很必要。
clust.br1 <- as.hclust(cut.fc$lower[[1]]) genes.br1 <- clust.br1$labels