
ML: 聚類算法R包-K中心點聚類
K-medodis與K-means比較相似,但是K-medoids和K-means是有區別的,不一樣的地方在於中心點的選取,在K-means中,我們將中心點取為當前cluster中所有數據點的平均值,在 K-medoids算法中,我們將從當前cluster 中選取這樣一個點——它到其他所有(當前cluster中的)點的距離之和最小——作為中心點。K-medodis算法不容易受到那些由於誤差之類的原因產生的臟數據的影響,但計算量顯然要比K-means要大,一般只適合小數據量。 K-medoids 主要運用到了R語言中cluster包中的pam函數
K中心點聚類
- cluster::pam
- fpc::pamk
cluster::pam
Usage: pam(x, k, diss = inherits(x, "dist"), metric = "euclidean", medoids = NULL, stand = FALSE, cluster.only = FALSE, do.swap = TRUE, keep.diss = !diss && !cluster.only && n < 100, keep.data = !diss && !cluster.only, pamonce = FALSE, trace.lev = 0)
- x:聚類對象
- k: 是聚類個數 ( positive integer specifying the number of clusters, less than the number of observations)
示例代碼
> newiris <- iris[,-5]
> library(cluster)
> kc <- pam(x=newiris,k=3)
> #kc$clustering
> #kc[1:length(kc)]
>
> table(iris$Species, kc$clustering)
小結:
針對K-均值算法易受極值影響這一缺點的改進算法.在原理上的差異在於選擇個類別中心點時不取樣本均值點,而在類別內選取到其余樣本距離之和最小的樣本為中心。
fpc::pamk
相比於pam函數,可以給出參考的聚類個數, 參考 kmenas 與 kmeansrun
Usage: pamk(data,krange=2:10,criterion="asw", usepam=TRUE, scaling=FALSE, alpha=0.001, diss=inherits(data, "dist"), critout=FALSE, ns=10, seed=NULL, ...)
示例代碼
newiris <- iris
newiris$Species <- NULL
library(fpc)
kc2 <- pamk(newiris,krang=1:5)
plot(pam(newiris, kc2$nc))
圖例
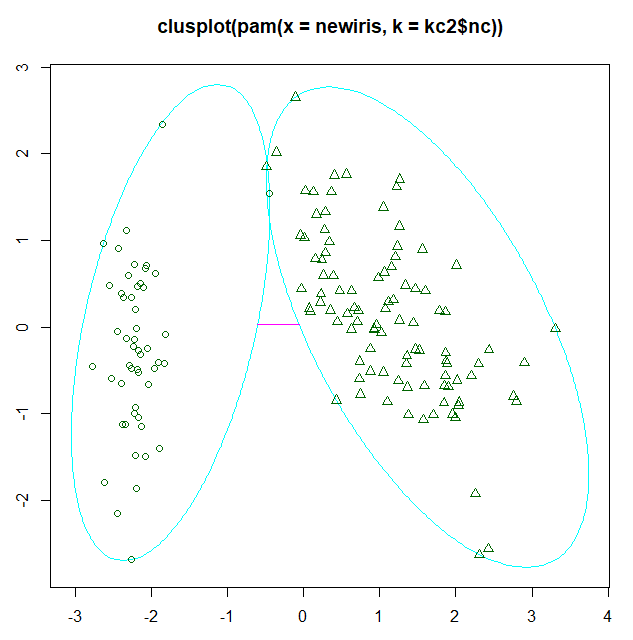
fpc包還提供了另一個展示聚類分析的函數plotcluster(),值得一提的是,數據將被投影到不同的簇中
plotcluster(newiris,kc2$cluster)
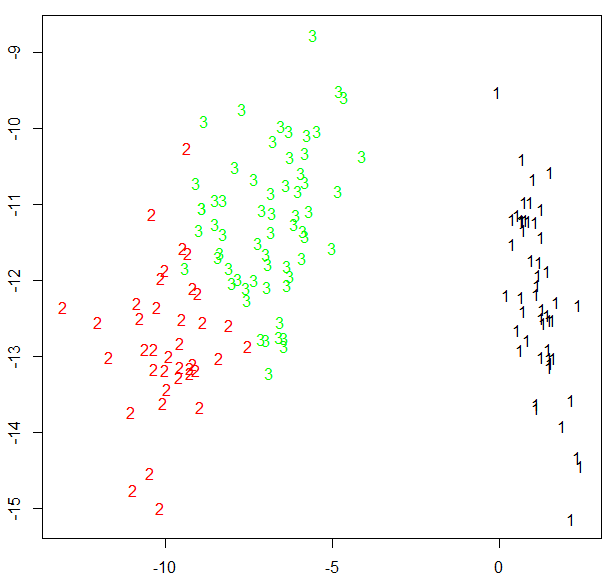
待驗證:
為什麽僅出現兩個聚類?
參考資料:
- http://blog.csdn.net/helen1313/article/details/38111125
ML: 聚類算法R包-K中心點聚類