
RNA-seq流程報告
實驗旨在瞭解RNA-seq的基本原理。通過模仿文獻《Targeting super enhancer associated oncogenes in oesophageal squamous cell carcinoma》的流程,學會利用NCBI和EBI資料庫下載資料,熟悉Linux下的基本操作,並使用R語言畫圖,用Python或者shell寫指令碼進行基本的資料處理,使用FastQC等軟體進行資料處理,學習Hisat2+StringTie+Ballgown的RNA-seq分析流程,並對結果進行分析討論。
1、硬體平臺
處理器:Intel(R) Core(TM)i7-4710MQ CPU @ 2.50GHz
安裝記憶體(RAM):16.0GB
2、系統平臺
Windows 8.1,Ubuntu
3、軟體平臺
① Aspera connect ② FastQC ③ Hisat2
④ StringTie ⑤ Ballgown
4、資料庫資源
NCBI資料庫:https://www.ncbi.nlm.nih.gov/;
EBI資料庫:http://www.ebi.ac.uk/
5、研究物件
加入H3K27Ac 抗體處理過的TE7細胞系測序資料和其空白對照組
加入H3K27Ac 抗體處理過的KYSE510細胞系和其空白對照組
背景簡介:食管鱗狀細胞癌(OSCC)是一種侵襲性的惡性腫瘤,本文章通過高通量小分子抑制劑進行篩選,發現了一個高度有效的抗癌物,特異性的CDK7抑制劑THZ1。RNA-Seq顯示,低劑量THZ1會對一些致癌基因的產生選擇性抑制作用,而且,對這些THZ1敏感的基因組功能的進一步表徵表明他們經常與超級增強子結合(SE)。Chip-seq解讀在OSCC細胞中,CDK7的抑制作用的機制。
本文亮點:確定了在OSCC細胞中SE的位置,以及識別出許多SE有關的調節元件;並且發現小分子THZ1特異性抑制SE有關的轉錄,顯示強大的抗癌性。
文章PMID: 27196599
1、Aspera軟體下載及安裝
進入Aspera官網的Downloads介面,選中aspera connect server,點選Wwindows圖示,選擇v3.6.2版本,點選Download進行下載。
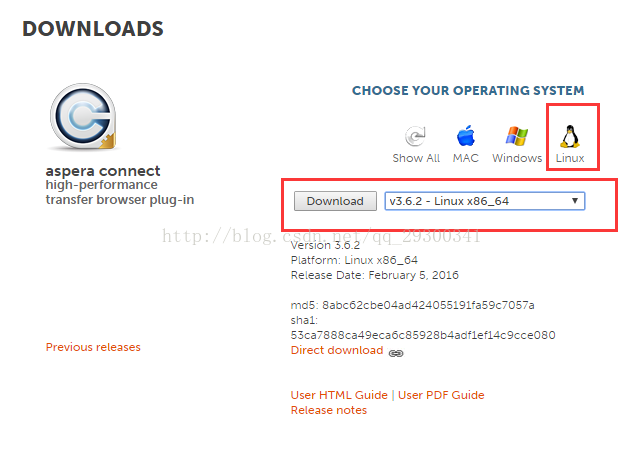
圖表 1 aspera的下載
Linux下的安裝配置參考博文:
http://blog.csdn.net/likelet/article/details/8226368
2、RNA-Seq資料下載
1)選擇NCBI的GEO DataSets資料庫,輸入GSE76861,開啟GSM2039114、GSM2039119、GSM2039120、GSM2039125獲取它們對應的SRX序列號。(此處僅僅做了KYSE510和TE7細胞系加入THZ1後0hr和12hr的RNA-seq資料)
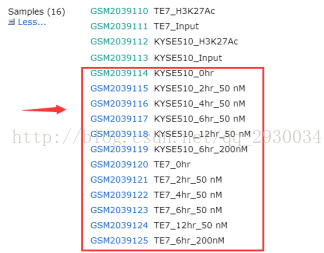
圖表 2 RNA-seq資料

圖表 3 獲取SRA編號
2)進入EBI,獲取ascp下載地址
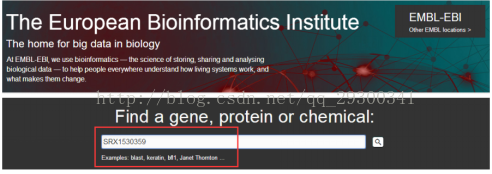
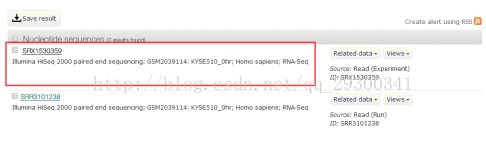

圖表 4 ascp下載地址
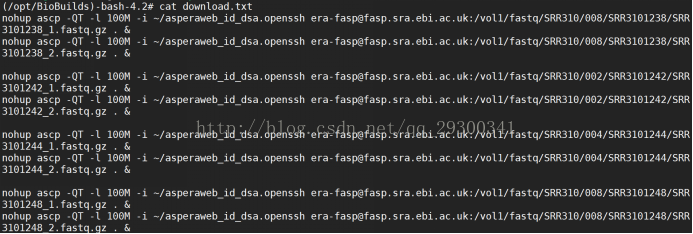
3)使用aspera下載並解壓
aspera下載命令及gunzip解壓命令(nohup+命令+& 可以後臺執行)
3、FastQC質量檢查
3.1 FastQC的安裝
Ubuntu軟體包內自帶Fastqc
故安裝命令apt-get install fastqc
3.2使用FastQC進行質量檢查
fastqc命令:
fastqc -o . -t 5 SRR3101238_1.fastq.gz &
-o . 將結果輸出到當前目錄
-t 5 表示開5個執行緒執行
(要分別對八個fastq檔案執行八次)
4、使用Hisat2對Reads進行Mapping
4.1 Hisat2的安裝
進入Hisat2官網http://ccb.jhu.edu/software/hisat2/index.shtml下載二進位制程式,再配置環境變數即可。
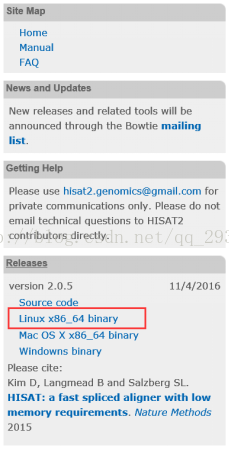
圖表 5 Hisat2下載與安裝
4.2下載人類參考基因組和註釋檔案
人類參考基因組:Hisat2官網上有Ensemble GRCh38的基因組索引,可以直接下載使用
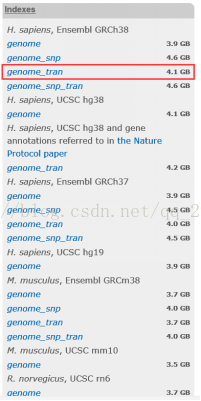
圖表 6 Hisat2建立的GRCh38索引
註釋檔案:下載自ensemble資料庫ftp://ftp.ensembl.org/pub/release-86/gtf/homo_sapiens
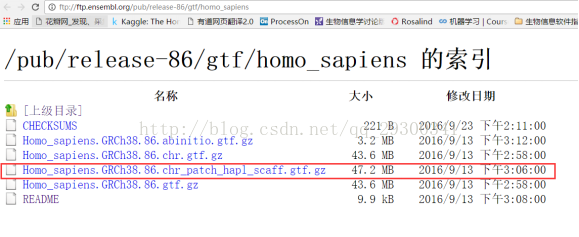
圖表 7 Ensemble註釋檔案
PS:如果是想自己下載基因組,然後自己建立索引,需要使用Hisat2包裡面的python指令碼
extract_splice_sites.py和extract_exons.py,從註釋檔案裡面抽取出剪下位點和外顯子資訊
First, using the python scripts included in the HISAT2 package, extract splice-site and exon information from the gene annotation file:
extract_splice_sites.py chrX_data/genes/chrX.gtf >chrX.ss
extract_exons.py chrX_data/genes/chrX.gtf >chrX.exon
Second, build a HISAT2 index:
hisat2-build --ss chrX.ss --exon chrX.exon chrX_data/genome/chrX.fa chrX_tran
The --ss and --exon options can be omitted in the command above if annotation is not available.
4.3 Hisat2比對
將RNA-seq的測序reads使用hisat2比對
5、samtools檔案格式轉化
samtools將sam檔案轉成bam,並且排序,為下游分析做準備
6、stringtie轉錄本處理
6.1stringtie組裝轉錄本
stringtie對每個樣本進行轉錄本組裝
6.2stringtie合併轉錄本
stringtie 將所有樣本的轉錄本進行合併
注意:此處的mergelist.txt是自己建立的,需要包含之前SRR3101238.gtf,SRR3101242.gtf,SRR3101244.gtf,SRR3101248.gtf的路徑
stringtie --merge -p 4 -G Homo_sapiens.GRCh38.86.chr_patch_hapl_scaff.gtf -o stringtie_merged.gtf mergelist.txt;
6.3 stringtie評估表達量
計算表達量並且為Ballgown包提供輸入檔案
7、Ballgown表達量分析
7.1 Ballgown的安裝
source("http://bioconductor.org/biocLite.R")
biocLite("ballgown")
7.2 檔案準備與分析
將資料的分組資訊寫入一個csv檔案,此處phenodata.csv檔案
phenodata.csv檔案內容
sampleid,celllines,time
SRR3101238,KYSE,0hr
SRR3101242,KYSE,12hr
SRR3101244,TE7,0hr
SRR3101248,TE7,12hr
接下來就可以用ballgown愉快地進行分析了。
此處只做了四個樣本,然後在Ballgown包裡用stattest是無法計算出P值的,樣本稍微多一些才可以檢驗。
1、RNA-Seq資料下載
RNA-Seq資料下載結果
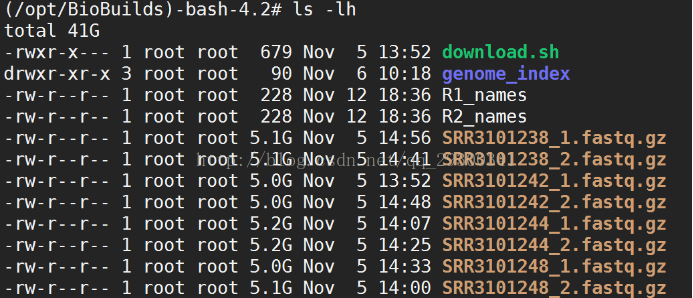
圖表 8 RNA-Seq資料
2、FastQC質量檢查
資料質量檢查
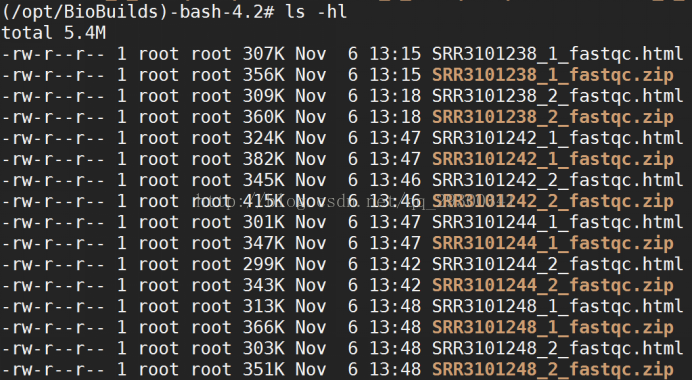
圖表 9 質量檢查檔案
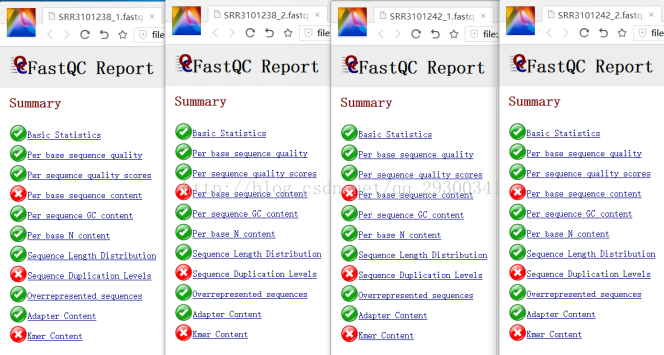
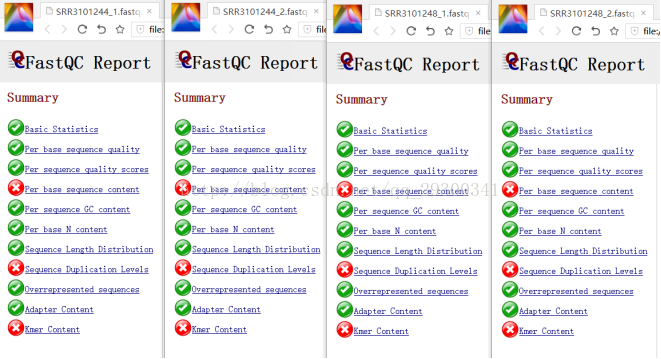
3、使用Hisat2對Reads進行Mapping
3.1基因組檔案
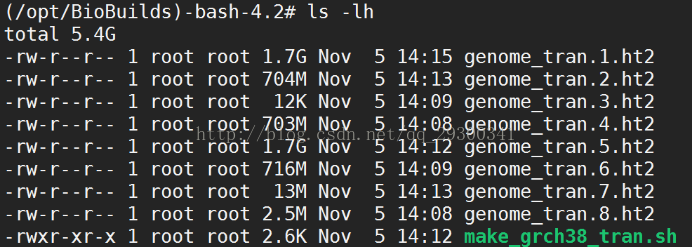
圖表 11人類參考基因組GRCh38索引
3.2 Mapping結果
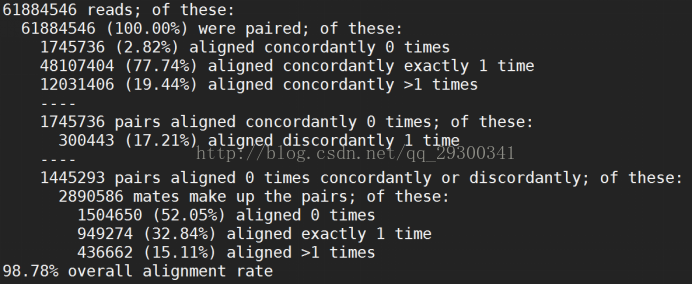
4、samtools結果
samtools結果檔案
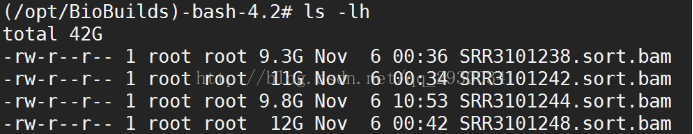
圖表 13檔案轉化結果
5、stringtie結果
5.1組裝轉錄本
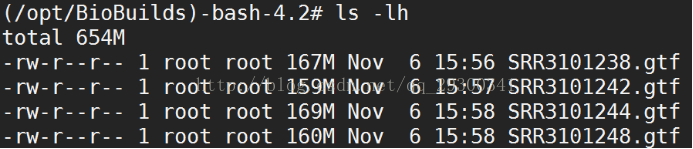
圖表 14 組裝轉錄本
5.2 合併轉錄本
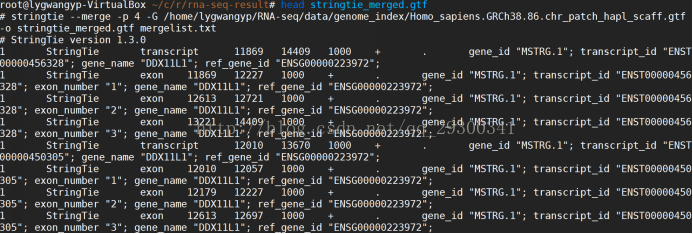
圖表 15合併轉錄本
5.3評估表達量
圖表 16評估表達量
6、Ballgown分析結果
剪下方式分析
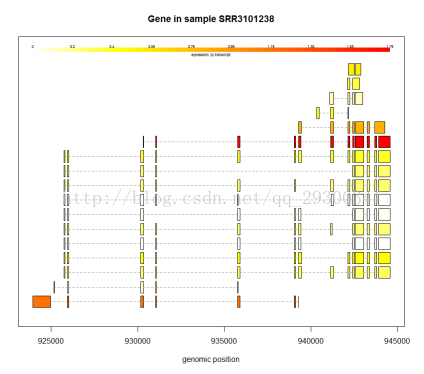
圖表 17 某個基因的剪下方式
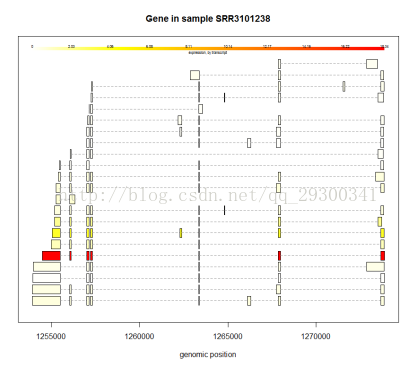
圖表 18某個基因的剪下方式
FastQC質量檢查
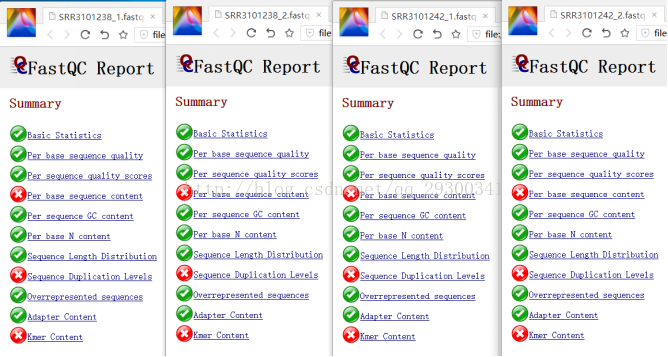
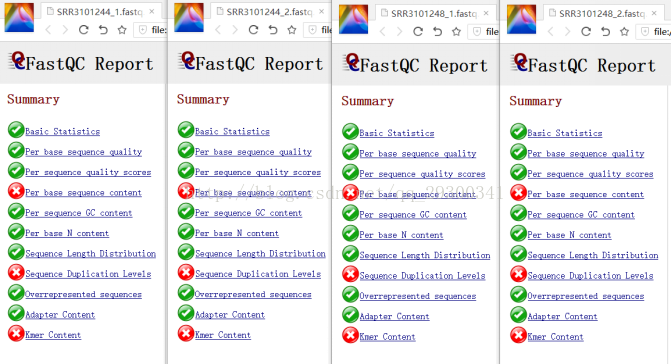
圖表 10 質量檢查結果顯示,測序質量挺好,Per base sequence content、Sequence Duplication Levels、Kmer Content出現警告更可能是由於測序方法本身存在的固有誤差。
Hisat2整體覆蓋度
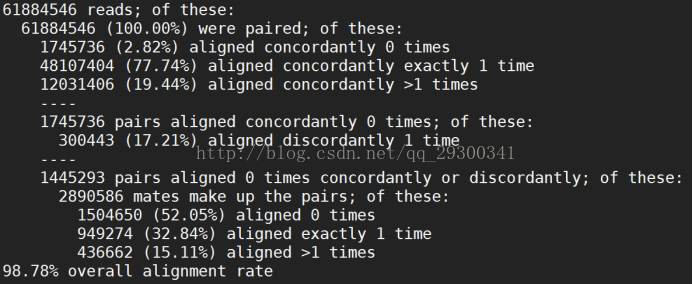
由圖表 12 Mapping整體結果可以看出,四個fastq檔案Mapping整體覆蓋率都在98%左右,從另一方面說明資料質量很好
可變剪下方式分析
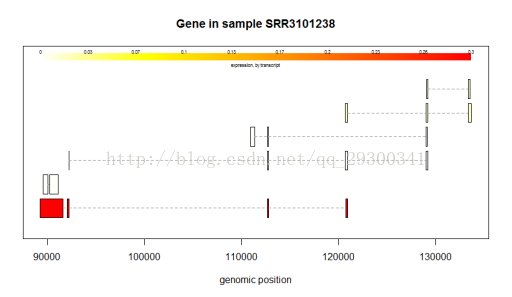
初步繪製了幾個基因的剪下方式圖,發現在可變剪下中,確實會有某一種方式佔據主導地位。
表達量熱力圖
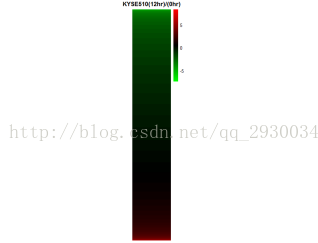

圖表 19 KYSE510細胞系和圖表 20 TE7細胞系的基因表達圖的計算方式為:
加藥12hr表達量÷0hr表達量,再取log2後的值;
由於表達量訊號值小於1不顯著,故篩去,僅留下訊號值都大於1的基因。
從圖中綠色較多,可以看出加入THZ1的12hr後,明顯抑制了基因的表達。
RNA-Seq與Chip-Seq結合分析
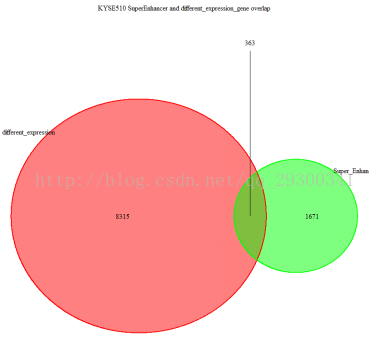
圖表 21 KYSE510結合分析顯示,KYSE510細胞系差異表達基因中,有363個與Chip-seq分析流程中鑑定出的SuperEnhancer相重疊。
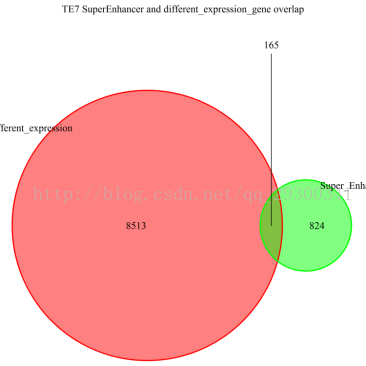
圖表 22 TE7結合分析顯示,TE7細胞系差異表達基因中,有165個與Chip-seq分析流程中鑑定出的SuperEnhancer相重疊。
Kobas註釋分析
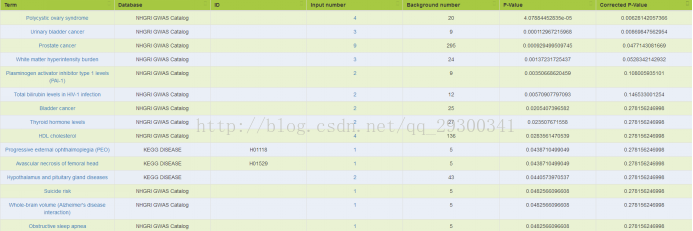
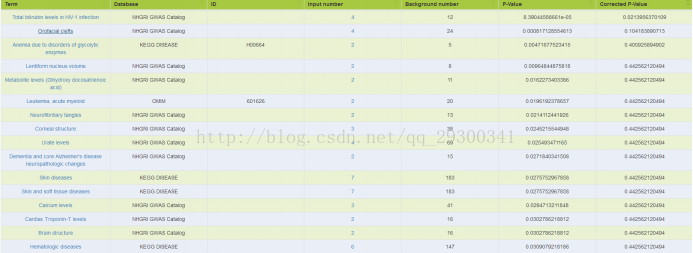
對於Chip-seq中SuperEnhancer和RNA-seq差異表達基因overlap的部分,先用Ensemble資料庫的Biomart,將GeneSymbol轉成EnsembleID,再輸入至Kobas註釋,註釋選用OMIM、KEGG DISEASE、NHGRI GWAS Catalog資料庫,由圖表 23 TE7結合分析基因註釋和圖表 24 KYSE510結合分析基因註釋結果來看,與癌症相關。
參考文獻
[1] Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma PMID: 27196599